Deep mutational scanning and machine learning reveal structural and molecular rules governing allosteric hotspots in homologous proteins
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
This article seeks to address a key question in protein biophysics: are the amino acid positions (and mutations) that influence allostery conserved across homologs of a protein family? Or is allostery implemented by a distinct set of residues that varies amongst homologs? To address this question, the authors follow an innovative approach that combines deep mutational scanning with machine learning. Significant revisions are required to clarify whether the conclusions of the study are well-supported by the data. The work is potentially highly relevant to protein engineers and biophysicists.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. The reviewers remained anonymous to the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
A fundamental question in protein science is where allosteric hotspots – residues critical for allosteric signaling – are located, and what properties differentiate them. We carried out deep mutational scanning (DMS) of four homologous bacterial allosteric transcription factors (aTFs) to identify hotspots and built a machine learning model with this data to glean the structural and molecular properties of allosteric hotspots. We found hotspots to be distributed protein-wide rather than being restricted to ‘pathways’ linking allosteric and active sites as is commonly assumed. Despite structural homology, the location of hotspots was not superimposable across the aTFs. However, common signatures emerged when comparing hotspots coincident with long-range interactions, suggesting that the allosteric mechanism is conserved among the homologs despite differences in molecular details. Machine learning with our large DMS datasets revealed global structural and dynamic properties to be a strong predictor of whether a residue is a hotspot than local and physicochemical properties. Furthermore, a model trained on one protein can predict hotspots in a homolog. In summary, the overall allosteric mechanism is embedded in the structural fold of the aTF family, but the finer, molecular details are sequence-specific.
Article activity feed
-

-

Author Response
Reviewer 1 (Public Review):
To me, the strengths of the paper are predominantly in the experimental work, there's a huge amount of data generated through mutagenesis, screening, and DMS. This is likely to constitute a valuable dataset for future work.
We are grateful to the reviewer for their generous comment.
Scientifically, I think what is perhaps missing, and I don't want this to be misconstrued as a request for additional work, is a deeper analysis of the structural and dynamic molecular basis for the observations. In some ways, the ML is used to replace this and I think it doesn't do as good a job. It is clear for example that there are common mechanisms underpinning the allostery between these proteins, but they are left hanging to some degree. It should be possible to work out what these are with further …
Author Response
Reviewer 1 (Public Review):
To me, the strengths of the paper are predominantly in the experimental work, there's a huge amount of data generated through mutagenesis, screening, and DMS. This is likely to constitute a valuable dataset for future work.
We are grateful to the reviewer for their generous comment.
Scientifically, I think what is perhaps missing, and I don't want this to be misconstrued as a request for additional work, is a deeper analysis of the structural and dynamic molecular basis for the observations. In some ways, the ML is used to replace this and I think it doesn't do as good a job. It is clear for example that there are common mechanisms underpinning the allostery between these proteins, but they are left hanging to some degree. It should be possible to work out what these are with further biophysical analysis…. Actually testing that hypothesis experimentally/computationally would be nice (rather than relying on inference from ML).
We agree with the reviewer that this study should motivate a deeper biophysical analysis of molecular mechanisms. However, in our view, the ML portion of our work was not intended as a replacement for mechanistic analysis, nor could it serve as one. We treated ML as a hypothesis-generating tool. We hypothesized that distant homologs are likely to have similar allosteric mechanisms which may not be evident from visual analysis of DMS maps. We used ML to (a) extract underlying similarities between homologs (b) make cross predictions across homologs. In fact, the chief conclusion of our work is that while common patterns exist across homologs, the molecular details differ. ML provides tantalizing evidence to this effect. The conclusive evidence will require, as the reviewer rightly suggests, detailed experimental or molecular dynamics characterization. Along this line, we note that we have recently reported our atomistic MD analysis of allostery hotspots in TetR (JACS, 2022, 144, 10870). See ref. 41.
Changes to manuscript:
“Detailed biophysical or molecular dynamics characterization will be required to further validate our conclusions(38).”Reviewer 3 (Public Review):
However - at least in the manuscript's present form - the paper suffers from key conceptual difficulties and a lack of rigor in data analysis that substantially limits one's confidence in the authors' interpretations.
We hope the responses below address and allay the reviewer’s concerns.
A key conceptual challenge shaping the interpretation of this work lies in the definition of allostery, and allosteric hotspot. The authors define allosteric mutations as those that abrogate the response of a given aTF to a small molecule effector (inducer). Thus, the results focus on mutations that are "allosterically dead". However, this assay would seem to miss other types of allosteric mutations: for example, mutations that enhance the allosteric response to ligand would not be captured, and neither would mutations that more subtly tune the dynamic range between uninduced ("off) and induced ("on") states (without wholesale breaking the observed allostery). Prior work has even indicated the presence of TetR mutations that reverse the activity of the effector, causing it to act as a co-repressor rather than an inducer (Scholz et al (2004) PMID: 15255892). Because the work focuses only on allosterically dead mutations, it is unclear how the outcome of the experiments would change if a broader (and in our view more complete) definition of allostery were considered.
We agree with the reviewer that mutations that impact allostery manifest in many different ways. Furthermore, the effect size of these mutations runs the full gamut from subtle changes in dynamic range to drastic reversal of function. To unpack allostery further, allostery of aTF can be described, not just by the dynamic range, but by the actual basal and induced expression levels of the reporter, EC50 and Hill coefficient. Given the systemic nature of allostery, a substantial fraction of aTF mutations may have some subtle impact on one or more of these metrics. To take the reviewer’s argument one step further, one would have to accurately quantify the effect size of every single amino acid mutation on all the above properties to have a comprehensive sequence-function landscape of allostery. Needless to say, this is extremely hard! Resolution of small effect sizes is very difficult, even at high sequencing depth. To the best of our knowledge, a heroic effort approaching such comprehensive analysis has been accomplished so far only once (PMID: 3491352).
Our focus, therefore, was to screen for the strongest phenotypic impact on allostery i.e., loss of function. Mutations leading to loss of function can be relatively easily identified by cell-sorting. Because our goal was to compare hotspots across homologs, we surmised that loss of function mutations, given their strong phenotypic impact, are likely to provide the clearest evidence of whether allosteric hotspots are conserved across remote homologs.
The reviewer raised the point of activity-reversing mutations. Yes, there are activity reversing mutations in TetR. However, they represent an insignificant fraction. In the paper cited by the reviewer, there are 15 activity-reversing mutations among 4000 screened. Furthermore, the paper shows that activity-reversing in TetR requires two-tofour mutations, while our library is exclusively single amino acid substitutions. For these reasons, we did not screen for activity-reversing mutations. Nonetheless, we agree with the reviewer that screening for activity-reversing mutations across homologs would be very interesting.
The separation in fluorescence between the uninduced and induced states (the assay dynamic range, or fold induction) varies substantially amongst the four aTF homologs. Most concerningly, the fluorescence distributions for the uninduced and induced populations of the RolR single mutant library overlap almost completely (Figure 1, supplement 1), making it unclear if the authors can truly detect meaningful variation in regulation for this homolog.
Yes, the reviewer is correct that the fold induction ratio varies among the four aTF homologs. However, we note that such differences are common among natural aTFs. Depending on the native downstream gene regulated by the aTF, some aTFs show higher ligand-induced activation, and others are lower. While this is not a hard and fast rule, aTFs that regulate efflux pumps tend to have higher fold induction than those that regulate metabolic enzymes. In summary, the variation in fold induction among the four aTFs is not a flaw in experimental design nor indicates experimental inconsistency but is instead just an inherent property of protein-DNA interaction strength and the allosteric response of each aTF.
Among the four aTFs, wildtype RolR has the weakest fold induction (15-fold) which makes sorting the RolR library particularly challenging. To minimize false positives as much as possible, we require that dead mutant be present in (a) non-fluorescent cells after ligandinduction (b) non-fluorescent cells before ligand-induction (c) at least two out of the three replicates for both sorts. Additionally, for RolR specifically, we adjusted the nonfluorescent gate to be far more stringent than the other three aTFs (Fig. 1 – figure supplement 1). Furthermore, we assign residues as allosteric hotspots, not individual dead mutations. This buffers against false strong signals from stray individual dead mutations. Finally, the top interquartile range winnows them to residues showing strong consistent dead phenotype. As a result of these “safeguards” we have built in, the number of allosteric hotspots of RolR (57) is comparable to the other three aTFs (51, 53 and 48). This suggests that we are not overestimating the number of hotspots despite the weaker fold induction of RolR. We highlight in a new supplementary figure (Figure 1 – figure supplement 4) that changing the read count threshold from 5X to 10X produces near identical patterns of mutations suggesting that our results are also robust to changes in ready depth stringency.
Changes to manuscript: In response to the reviewer's comment, we have added the following sentence.
“We note that the lower fold induction (dynamic range) of RolR makes it particularly challenging to separate the dead variants from the rest.”
The methods state that "variants with at least 5 reads in both the presence and absence of ligand in at least two replicates were identified as dead". However, the use of a single threshold (5 reads) to define allosterically dead mutations across all mutations in all four homologs overlooks several important factors:
Depending on the starting number of reads for a given mutation in the population (which may differ in orders of magnitude), the observation of 5 reads in the gated nonfluorescent region might be highly significant, or not significant at all. Often this is handled by considering a relative enrichment (say in the induced vs uninduced population) rather than a flat threshold across all variants.
We regret the lack of clarity in our presentation. We wish to better explain the rationale behind our approach. First, we understand the reviewer’s point on considering relative enrichment to define a threshold. This approach works well in DMS experiments involving genetic selections, which is commonly the case, because activity scales well with selection stringency. One can then pick enrichment/depletion relative to the middle of the read count distribution as a measure of gain or loss of function.
Second, this strategy does not, in practice, work well for cell-sorting screens. While it may be tempting to think of cell sorting as comparably activity-scaled as genetic selections, in reality, the fidelity of fluorescent-activated cell sorters is much lower. Making quantitative claims of activity based on cell sorting enrichment can be risky. It is wiser to treat cell sorting results as yes/no binary i.e., does the mutation disrupt allostery or not. More importantly, the yes/no binary classification suffices for our need to identify if a certain mutation adversely impacts allosteric activity or not.
Third, the above argument does not imply that all mutations have the same effect size on allostery. They don’t. We capture the effect size on individual residues, not individual mutations, by counting the number of dead mutations at a residue position. This is an important consideration because it safeguards us from minor inconsistencies that inevitably arise from cell sorting.
Fourth, a variant to be classified as allosterically dead, it must be present both in uninduced and induced DNA-bound populations in at least two out of three replicates (four conditions total). This is a stringent criterion for selecting dead variants resulting in highly consistent regions of importance in the protein even upon varying read count thresholds. To the extent possible, we have minimized the possibility of false positive bleed-through.
Finally, two separate normalizations were performed on the total sequence reads to be able to draw a common read count threshold 1) between experimental conditions & replicates and 2) across proteins. First, total sequencing reads were normalized to 200k total across all sample conditions (presorted, -inducer, and +inducer) and replicates for each homolog, allowing comparisons within a single protein. Next, reads were normalized again to account for differences in the theoretical size of each protein’s single-mutant library, allowing for comparisons across proteins by drawing a commont readcount cutoff. For example, total sequencing reads of RolR (4,332 possible mutants) increased by 1.18x relative to MphR (3,667 possible mutants) for a total of 236k reads.
Changes to manuscript: We have provided substantial additional details in the Fluorescence-activated cell sorting and NGS preparation and analysis sections.
We also added the following in the main text.
“In other words, we use cell sorting as a binary classifier i.e., does the mutation disrupt allostery or not. We capture the effect size on individual residues, not individual mutations, by counting the number of dead mutations at a residue position. This is an important consideration because it safeguards us from minor inconsistencies that inevitably arise from cell sorting.”
Depending on the noise in the data (as captured in the nucleotide-specific q-scores) and the number of nucleotides changed relative to the WT (anywhere between 1-3 for a given amino acid mutation) one might have more or less chance of observing five reads for a given mutation simply due to sequencing noise.
All the reads considered in our analyses pass the Illumina quality threshold of Q-score ≥ 30 which as per Illumina represent “perfect reads with no errors or ambiguities”. This translates into a probability of 1 in 1000 incorrect base call or 99.9% base call accuracy.
We use chip-based oligonucleotides to build our DMS library, which allows us to prespecify the exact codon that encodes a point mutation. This means the nucleotide count and protein count are the same. The scenario referred to by the reviewer i.e., “anywhere between 1-3 for a given amino acid mutation” only applies to codon randomized or errorprone PCR library generation. We regret if the chip-based library assembly part was unclear.
Depending on the shape and separation of the induced (fluorescent) and uninduced (non-fluorescent) population distributions, one might have more or less chance of observing five reads by chance in the gated non-fluorescent region. The current single threshold does not account for variation in the dynamic range of the assay across homologs.
We have addressed the concern raised by the reviewer on fluorescent population distributions in answers to questions 10 and 11.
The reviewer makes an important point about the choice of sequencing threshold. We use the sequencing threshold to simply make a binary choice for whether a certain variant exists in the sorted population or not. We do not use the sequencing reads as to scale the activity of the variant. To address the reviewer's comment, we have included a new supplementary figure (Fig 1 – figure supplement 4) where we compare the data by adjust the threshold two levels – 5 and 10 reads. As is evident in the new figure, the fundamental pattern of allosteric hotspots and the overall data interpretation does not change.
TetR: 5x – 53 hotspots, 10x – 51 hotspots
TtgR: 5x – 51 hotspots, 10x – 51 hotspots
MphR: 5x – 48 hotspots, 10x – 48 hotspots
RolR: 5x – 57 hotspots, 10x – 60 hotspots
In other words, changing the threshold to be more or less strict may have a modest impact on the overall number of hotspots in the dataset. Still, the regions of functional importance are consistent across different thresholds. We have expanded the discussion in the manuscript to address this point.
Changes to manuscript: We have now included a new supplementary comparing hotspot data at two thresholds: Figure 1 – figure supplement 4.
We also added the following in the main text.
“To assess the robustness of our classification of hotspots, we determined the number of hotspots at two different sequencing thresholds – 5x and 10x. At 5x and 10x, the number of hotspots are – TetR: 53, 51; TtgR: 51, 51; MphR: 48, 48 and RolR: 57,60, respectively. Changing the threshold has a modest impact on the overall number of hotspots and the regions of functional importance are consistent at both thresholds”
The authors provide a brief written description of the "weighted score" used to define allosteric hotspots (see y-axis for figure 1B), but without an equation, it is not clear what was calculated. Nonetheless, understanding this weighted score seems central to their definition of allosteric hotspots.
We regret the lack of clarity in our presentation. The weighted score was used to quantify the “deadness” of every residue position in the protein. At each position in the protein, the number of mutations that inhibited activity was summed up and the ‘deadness’ of each mutation was weighted based on how many replicates is appeared to inactivate the protein. Weighted score at each residue position is given by
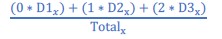
Where at position x in the protein, D1 is the number of mutations dead in one replicate only, D2 is the number of mutations dead in 2 replicates, D3 is the number of mutations dead in 3 replicates, and Total is the total number of variants present in the data set (based on sequencing data). Any dead mutation that is seen in only one replicate is discarded and does not contribute to the “deadness” of the residue. Mutations seen in two and three replicates contribute to the score. We have included a new supplementary figure (Fig. 1 – figure supplement 2) to give the reader a detailed heatmap of all mutations and their impact for each protein.
Changes to manuscript: The weighted scoring scheme is now described in greater detail under Materials and Methods in the “NGS preparation and analysis” section.
The authors do not provide some of the standard "controls" often used to assess deep mutational scanning data. For example, one might expect that synonymous mutations are not categorized as allosterically dead using their methods (because they should still respond to ligand) and that most nonsense mutations are also not allosterically dead (because they should no longer repress GFP under either condition). In general, it is not clear how the authors validated the assay/confirmed that it is giving the expected results.
As we state in response to question 12, we use chip-based oligonucleotides to build our DMS library, which allows us to pre-specify the exact codon that encodes a point mutation. We have no synonymous or nonsense mutations in our DMS library. Each protein mutation is encoded by a single unique codon. The only stop codon is at 3’end of the gene.
The authors performed three replicates of the experiment, but reproducibility across replicates and noise in the assay is not presented/discussed.
Changes to manuscript: A new supplementary table (Table 1) is now provided with the pairwise correlation coefficients between all replicates for each protein.
In the analysis of long-range interactions, the authors assert that "hotspot interactions are more likely to be long-range than those of non-hotspots", but this was not accompanied by a statistical test (Figure 2 - figure supplement 1).
In response to the reviewer's comment, we now include a paired t-test comparing nonhotspots and hotspots with long-range interactions in the main text.
Changes to manuscript: In all four aTFs, hotspots constituted a higher fraction of LRIs than non-hotspots (Figure 2 – figure supplement 1; P = 0.07).
-
Evaluation Summary:
This article seeks to address a key question in protein biophysics: are the amino acid positions (and mutations) that influence allostery conserved across homologs of a protein family? Or is allostery implemented by a distinct set of residues that varies amongst homologs? To address this question, the authors follow an innovative approach that combines deep mutational scanning with machine learning. Significant revisions are required to clarify whether the conclusions of the study are well-supported by the data. The work is potentially highly relevant to protein engineers and biophysicists.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. The reviewers remained anonymous to the authors.)
-
Reviewer #1 (Public Review):
This is an interesting paper, which has used cutting-edge approaches (DMS and ML) to probe an important phenomenon in protein function, namely allostery. The paper managed to acquire a large volume of data and to use this data efficiently to train ML models, which are then used to probe the question of why are some regions "allosteric" hot spots. The results are interesting and novel and suggest that despite structural homology, hotspot regions can differ among relatively close relatives, nevertheless, there are common mechanisms underpinning the allosteric mechanisms, likely linked to the conformational sampling of the proteins.
Strengths - To me, the strengths of the paper are predominantly in the experimental work, there's a huge amount of data generated through mutagenesis, screening, and DMS. This is …
Reviewer #1 (Public Review):
This is an interesting paper, which has used cutting-edge approaches (DMS and ML) to probe an important phenomenon in protein function, namely allostery. The paper managed to acquire a large volume of data and to use this data efficiently to train ML models, which are then used to probe the question of why are some regions "allosteric" hot spots. The results are interesting and novel and suggest that despite structural homology, hotspot regions can differ among relatively close relatives, nevertheless, there are common mechanisms underpinning the allosteric mechanisms, likely linked to the conformational sampling of the proteins.
Strengths - To me, the strengths of the paper are predominantly in the experimental work, there's a huge amount of data generated through mutagenesis, screening, and DMS. This is likely to constitute a valuable dataset for future work. The experimental data allows mapping of the hotspots and much of the paper would be the same in terms of analysis without the ML, I think the experimental work with structural and sequence analysis would probably constitute a complete and impactful study alone, such is the quality. The ML obviously adds another layer of insight into the project. What is shown is that training on one homolog can allow the prediction of hotspots on related homologs. To some degree, this is as expected given these proteins share a common fold and function, yet the fact it is possible (albeit imperfect) despite quite a low sequence identity is notable.
Weaknesses - it is hard to describe this as a weakness, but the ML is obviously not perfect in the predictions, yet is still interesting. I don't have any major suggestions for revisions or changes - it is what it is and I think serves as a nice benchmark for follow-up studies with new methods and approaches. I think this reiterates the importance that the raw data is made available so that it can be used to benchmark alternative approaches and help advance the field. Scientifically, I think what is perhaps missing, and I don't want this to be misconstrued as a request for additional work, is a deeper analysis of the structural and dynamic molecular basis for the observations. In some ways, the ML is used to replace this and I think it doesn't do as good a job. It is clear for example that there are common mechanisms underpinning the allostery between these proteins, but they are left hanging to some degree. It should be possible to work out what these are with further biophysical analysis. To me, it is clear what we see here is likely some conservation in the dynamics of these proteins across the superfamily, and the allosteric mechanism involves modulation of the conformational sampling - which can happen through mutations/binding at different regions. Actually testing that hypothesis experimentally/computationally would be nice (rather than relying on inference from ML).
Achievement of aims: I think the aims are achieved, with the caveat as mentioned above, that the molecular basis for the observations is not really investigated or tested. The results support many of the conclusions, but without biophysical analysis, there is unavoidably some speculation in the discussion (which is reasonable and fine).
Impact: I think this will be impactful. I am sure others will love to get their hands on the data to run their own ML studies on, and the conclusions are interesting and impactful (seeing "deep" shared allostery across a fold). I think it is consistent with our understanding that protein folds have deep shared conformational tendencies, and that conformational sampling is at the core of much of what we term allostery.
-
Reviewer #2 (Public Review):
In this paper, the authors used deep mutational scanning (DMS) with statistical analysis to identify allosteric hotspots by systematically dissecting the functional contribution of each residue to allosteric signaling, providing a new way of identifying and probing allosteric hotspots by DMS. They observe that hotspot residues are distributed across the structure rather than being limited to the specific pathway(s) that connect the inducer and DNA binding sites as commonly assumed in allostery models.
Specifically, here the authors aim to predict allosteric residues. They also ask whether they are conserved among homologous proteins. Toward these goals, they combine deep mutational screening and machine learning analyses of four homologous aTFs. They observe that the hotspots distribution and the parameters …
Reviewer #2 (Public Review):
In this paper, the authors used deep mutational scanning (DMS) with statistical analysis to identify allosteric hotspots by systematically dissecting the functional contribution of each residue to allosteric signaling, providing a new way of identifying and probing allosteric hotspots by DMS. They observe that hotspot residues are distributed across the structure rather than being limited to the specific pathway(s) that connect the inducer and DNA binding sites as commonly assumed in allostery models.
Specifically, here the authors aim to predict allosteric residues. They also ask whether they are conserved among homologous proteins. Toward these goals, they combine deep mutational screening and machine learning analyses of four homologous aTFs. They observe that the hotspots distribution and the parameters which are involved are global, with the hotspots present throughout the structure, rather than along allosteric signaling pathways, which has been the classical premise. They also observe that there is no apparent direct structural link of the hotspots to either the allosteric or the active site and that they may occupy different positions in homologous proteins.
The methods, the observations, and the concept are highly innovative and present a new view of allostery. Considering that allostery is of fundamental importance in cell life, with key roles in practically all regulation mechanisms of protein actions, and is increasingly important in drug discovery, this work is of fundamental importance.
It is clear that a vast amount of work has been carried out in this combined experimental/computational study.
-
Reviewer #3 (Public Review):
Leander et al used deep mutational scanning to assess the effect of nearly all possible point mutations on four homologous bacterial allosteric transcription factors (aTFs). In particular, they identified mutations that abrogated the transcription factor response to a small molecule effector. The authors go on to use machine learning to determine which physicochemical properties distinguish mutations with allostery-eliminating effects from those without an effect. They report that mutations that eliminate the allosteric response to small molecules are quite variable across homologs and that global features are more predictive of which mutations will break allostery relative to local properties. Overall, the experimental strategy is well-chosen, and a comprehensive comparison of mutational sensitivity across …
Reviewer #3 (Public Review):
Leander et al used deep mutational scanning to assess the effect of nearly all possible point mutations on four homologous bacterial allosteric transcription factors (aTFs). In particular, they identified mutations that abrogated the transcription factor response to a small molecule effector. The authors go on to use machine learning to determine which physicochemical properties distinguish mutations with allostery-eliminating effects from those without an effect. They report that mutations that eliminate the allosteric response to small molecules are quite variable across homologs and that global features are more predictive of which mutations will break allostery relative to local properties. Overall, the experimental strategy is well-chosen, and a comprehensive comparison of mutational sensitivity across allosteric homologs is highly important to understand how conserved (or not) the implementation of allostery is across homologs. Moreover, the idea to use machine learning to assess which features are most predictive of "allosteric hotspots" is very nice, and provides some insight into what physical properties distinguish mutations that influence allostery. The authors include some interesting results on transfer learning (evaluating whether models trained on one protein predict allostery in another), and the use of alternate sequence representations (e.g. UniRep) in their machine learning analyses. However - at least in the manuscript's present form - the paper suffers from key conceptual difficulties and a lack of rigor in data analysis that substantially limits one's confidence in the authors' interpretations. More specifically:
A key conceptual challenge shaping the interpretation of this work lies in the definition of allostery, and allosteric hotspot. The authors define allosteric mutations as those that abrogate the response of a given aTF to a small molecule effector (inducer). Thus, the results focus on mutations that are "allosterically dead". However, this assay would seem to miss other types of allosteric mutations: for example, mutations that enhance the allosteric response to ligand would not be captured, and neither would mutations that more subtly tune the dynamic range between uninduced ("off) and induced ("on") states (without wholesale breaking the observed allostery). Prior work has even indicated the presence of TetR mutations that reverse the activity of the effector, causing it to act as a co-repressor rather than an inducer (Scholz et al (2004) PMID: 15255892). Because the work focuses only on allosterically dead mutations, it is unclear how the outcome of the experiments would change if a broader (and in our view more complete) definition of allostery were considered.
The experimental determination of which mutations impacted allostery is given only a limited description in the methods, but if we understand what was done, the analysis seems to neglect both (1) important caveats due to assay specifics and (2) more general limitations of deep mutational scanning data. In particular:
a. The separation in fluorescence between the uninduced and induced states (the assay dynamic range, or fold induction) varies substantially amongst the four aTF homologs. Most concerningly, the fluorescence distributions for the uninduced and induced populations of the RolR single mutant library overlap almost completely (Figure 1, supplement 1), making it unclear if the authors can truly detect meaningful variation in regulation for this homolog.
b. The methods state that "variants with at least 5 reads in both the presence and absence of ligand in at least two replicates were identified as dead". However, the use of a single threshold (5 reads) to define allosterically dead mutations across all mutations in all four homologs overlooks several important factors:
i. Depending on the starting number of reads for a given mutation in the population (which may differ in orders of magnitude), the observation of 5 reads in the gated non-fluorescent region might be highly significant, or not significant at all. Often this is handled by considering a relative enrichment (say in the induced vs uninduced population) rather than a flat threshold across all variants.
ii. Depending on the noise in the data (as captured in the nucleotide-specific q-scores) and the number of nucleotides changed relative to the WT (anywhere between 1-3 for a given amino acid mutation) one might have more or less chance of observing five reads for a given mutation simply due to sequencing noise.
iii. Depending on the shape and separation of the induced (fluorescent) and uninduced (non-fluorescent) population distributions, one might have more or less chance of observing five reads by chance in the gated non-fluorescent region. The current single threshold does not account for variation in the dynamic range of the assay across homologs.
c. The authors provide a brief written description of the "weighted score" used to define allosteric hotspots (see y-axis for figure 1B), but without an equation, it is not clear what was calculated. Nonetheless, understanding this weighted score seems central to their definition of allosteric hotspots
d. The authors do not provide some of the standard "controls" often used to assess deep mutational scanning data. For example, one might expect that synonymous mutations are not categorized as allosterically dead using their methods (because they should still respond to ligand) and that most nonsense mutations are also not allosterically dead (because they should no longer repress GFP under either condition). In general, it is not clear how the authors validated the assay/confirmed that it is giving the expected results.In several places, the manuscript lacks important statistical analyses needed to firmly establish the authors' claims
a. The authors performed three replicates of the experiment, but reproducibility across replicates and noise in the assay is not presented/discussed
b. In the analysis of long-range interactions, the authors assert that "hotspot interactions are more likely to be long-range than those of non-hotspots", but this was not accompanied by a statistical test (Figure 2 - figure supplement 1)Data availability and analysis transparency need improvement. The raw fastq reads do not seem to be publicly available, nor did we see access to the code used to perform the analysis. If the code is not provided, the description of the analysis in the methods section needs to be more detailed for reproducibility.
Overall, these concerns with fundamental aspects of the data analysis make it challenging to assess the reproducibility of the results, the fidelity of the assay (in reporting allosterically dead mutations), and the extent to which the data robustly support the authors' claims.
-
