Molecular mechanism of Afadin substrate recruitment to the receptor phosphatase PTPRK via its pseudophosphatase domain
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
These studies establish a role for the D2 pseudophosphatase domain of the PTPRK receptor-like phosphotyrosine phosphatase in recruiting Afadin, a cell-cell junction protein that is reported to be a PTPRK substrate, for dephosphorylation by the active D1 phosphatase domain. These findings suggest that the D2 pseudophosphatase domains of RPTPKs might have a general function as platforms to recruit specific phosphotyrosine substrates.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 and Reviewer #3 agreed to share their name with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Protein tyrosine phosphatase receptor-type kappa (PTPRK) is a transmembrane receptor that links extracellular homophilic interactions to intracellular catalytic activity. Previously we showed that PTPRK promotes cell–cell adhesion by selectively dephosphorylating several cell junction regulators including the protein Afadin (Fearnley et al, 2019). Here, we demonstrate that Afadin is recruited for dephosphorylation by directly binding to the PTPRK D2 pseudophosphatase domain. We mapped this interaction to a putative coiled coil (CC) domain in Afadin that is separated by more than 100 amino acids from the substrate pTyr residue. We identify the residues that define PTP specificity, explaining how Afadin is selectively dephosphorylated by PTPRK yet not by the closely related receptor tyrosine phosphatase PTPRM. Our work demonstrates that PTP substrate specificity can be determined by protein–protein interactions distal to the active site. This explains how PTPRK and other PTPs achieve substrate specificity despite a lack of specific sequence context at the substrate pTyr. Furthermore, by demonstrating that these interactions are phosphorylation-independent and mediated via binding to a non-catalytic domain, we highlight how receptor PTPs could function as intracellular scaffolds in addition to catalyzing protein dephosphorylation.
Article activity feed
-

-

Author Response
Reviewer #3 (Public Review):
- Validation of reagents: The authors generated a pY1230 Afadin antibody claiming that (page 6) "this new antibody is specific to tyrosine phosphorylated Afadin, and that pY1230 is targeted for dephosphorylation by PTPRK, in a D2-domain dependent manner". The WB in Fig 1B shows a lot of background, two main bands are visible which both diminish in intensity in ICT WT pervanadate-treated MCF10A cell lysates. The claim that the developed peptide antibody is selective for pY1230 in Afadin would need to be substantiated, for instance by pull down studies analysed by pY-MS to substantiate a claim of antibody specificity for this site. However, for the current study it would be sufficient to demonstrate that pY1230 is indeed the dephosphorylated site. I suggest therefore including a site directed …
Author Response
Reviewer #3 (Public Review):
- Validation of reagents: The authors generated a pY1230 Afadin antibody claiming that (page 6) "this new antibody is specific to tyrosine phosphorylated Afadin, and that pY1230 is targeted for dephosphorylation by PTPRK, in a D2-domain dependent manner". The WB in Fig 1B shows a lot of background, two main bands are visible which both diminish in intensity in ICT WT pervanadate-treated MCF10A cell lysates. The claim that the developed peptide antibody is selective for pY1230 in Afadin would need to be substantiated, for instance by pull down studies analysed by pY-MS to substantiate a claim of antibody specificity for this site. However, for the current study it would be sufficient to demonstrate that pY1230 is indeed the dephosphorylated site. I suggest therefore including a site directed mutant (Y1230F) that would confirm dephosphorylation at this site and the ability of the antibody recognizing the phosphorylation state at this position.
We would like this antibody to be a useful and freely accessible tool in the field and have taken on board the request for additional validation. To this end we have significantly expanded Supplementary Figure 2 (now Figure 1 - figure supplement 2) and included a dedicated section of the results as follows:
- We have now included information about all of the Afadin antibodies used in this study, since Afadin(BD) appears to be sensitive to phosphorylation (Figure 1 - figure supplement 2A).
- We have demonstrated that the Afadin pY1230 antibody detects an upregulated band in PTPRK KO MCF10A cells, consistent with our previous tyrosine phosphoproteomics (Figure 1 - figure supplement 2B). This indicates that the antibody can be used to detect endogenous Afadin phosphorylation.
- We have included two new knock down experiments demonstrating the recognition of Afadin by our antibody (Figure 1 - figure supplement 2C). There appear to be two Afadin isoforms recognised in HEK293T cells by both the BD and pY1230 antibody, consistent with previous reports (Umeda et al. MBoC, 2015). We have highlighted these in the figure.
- We have performed mutagenesis to demonstrate the specificity of the antibody. We tagged Afadin with a fluorescent protein tag, reasoning that it would cause a shift in molecular weight that could be resolved by SDS PAGE, as is the case. We noted that the phosphopeptide used spans an additional tyrosine, Y1226, which has been detected as phosphorylated (although to a much lower extent than Y1230) on Phosphosite plus. The data clearly show that Afadin cannot be phosphorylated when Y1230 is mutated to a phenylalanine (compared to CIP control), indicating that this is the predominant site recognised by the antibody. In addition, the endogenous pervanadate-stimulated signal is completely abolished by CIP treatment (Figure 1 - figure supplement 2D).
- We have included densitometric quantification of the dephosphorylation assay shown in Figure 1B, which was part of a time course and shows preferential dephosphorylation by the PTPRK ICD compared to the PTPRK D1. The signal stops declining with time, which could indicate antibody background, or an inaccessible pool of Afadin-pY1230 (Figure 1 - figure supplement 2E).
- To further demonstrate that this site is modulated by PTPRK in post-confluent cells, we have used doxycycline (dox)-inducible cell lines generated in Fearnley et al, 2019. Upon treatment with 500 ng/ml Dox for 48 hours PTPRK is induced to lower levels than wildtype, however, normalized quantification of the Afadin pY1230 against the Afadin (CST) signal clearly indicates downregulation by PTPRK WT, but not the catalytically inactive mutant (Figure 1 - figure supplement 2F and 2G). Together these data strengthen our assertion that this antibody recognises endogenously phosphorylated Afadin at site Y1230, which is modulated in vitro and in cells by PTPRK phosphatase activity. For clarity, we have highlighted and annotated the relevant bands in figures. We have also included identifiers for each Afadin total antibody was used in particular experiments.
- The authors claim that a short, 63-residue predicted coiled coil (CC) region, is both necessary and sufficient for binding to the PTPRK-ICD. The region is predicted to have alpha-helical structure and as a consequence, a helical structure has been used in the docking model. Considering that the authors recombinantly expressed this region in bacteria, it would be experimentally simple confirming the alpha-helical structure of the segment by CD or NMR spectroscopy.
To clarify, the helical structure in the docking model was independently predicted by several sequence and structural analysis programmes including AlphaFold2, RobettaFold, NetSurfP and as annotated in Uniprot (as a coiled coil). We did not stipulate prior to the AF2 prediction that it was helical. Isolated short peptides frequently adopt helical structure, therefore prediction of a helix within the context of the full Afadin sequence is, in our opinion, stronger evidence than CD of an isolated fragment.
- Only two mutants have been introduced into PTPRK-ICD to map the Afadin interaction site. One of the mutations changes a possibly structurally important residues (glycine) into a histidine. Even though this residue is present in PTPRM, it does not exclude that the D2 domain no longer functionally folds. Also the second mutation represents a large change in chemical properties and the other 2 predicted residues have not been investigated.
The residues that were selected for mutation are all localised to the protein surface and therefore are unlikely to be involved in stable folding of PTPRK. In support of the correct folding of the mutated PTPRK, we include in Figure 1 below SEC elution traces for wild-type and mutant D2 showing that they elute as single symmetric peaks at the same elution volume as the WT protein. This is consistent with them having a similar shape and size, and not being aggregated or unfolded.
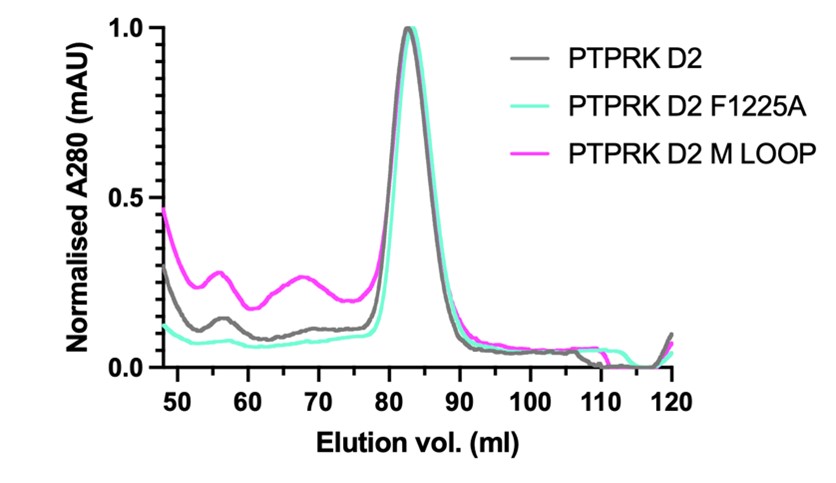
Figure 1. PTPRK-D2 wild-type and mutant preparative SEC elution profiles. A280nm has been normalised to help illustrate that the different proteins elute at the same volume. The main peak from these samples was used for binding assays in the main paper.
Furthermore, the yield for the double mutant was very high (4 mg of pure protein from a 2 L culture, see A280 value in graph below), whereas poorly folded proteins tend to have significantly reduced yields. This protein was also very stable over time whereas unfolded proteins tend to degrade during or following purification.
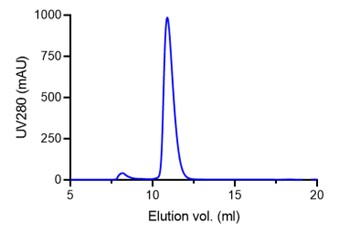
Figure 2. Analytical SEC elution profile for the PTPRK-D2 DM construct showing the very high yield consistent with a well-folded, stable protein.
Finally, we have carried out thermal melt curves of the WT and mutant PTPRK D2 domains showing that they all possess melting temperatures between 39.3°C and 41.7°C, supporting that they are all equivalently folded. We include these data as an additional Supplementary Figure (Figure 4 - figure supplement 3) in the paper.
- The interface on the Afadin substrate has not been investigated apart from deleting the entire CC or a central charge cluster. Based on the docking model the authors must have identified key positions of this interaction that could be mutated to confirm the proposed interaction site.
We have now made and tested several additional mutations within both the Afadin-CC and PTPRK-D2 domains to further validate the AF2 predicted model of the complex.
For Afadin-CC we introduced several single and double mutations along the helix including residues predicted to be in the interface and residues distal from the interface. These mutations and the pulldown with PTPRK are described in the text and are included as additional panels to a modified Figure 3. All mutations have the expected effect on the interaction based on the predicted complex structure. To help illustrate the positions of these mutations we have also included a figure of the interface with the residues highlighted.
For the PTPRK-D2 we have also introduced two new mutations, one buried in the interface (F1225A) and one on the edge of the interface encompassing a loop that is different in PTPRM (labelled the M-loop). GST-Afadin WT protein was bound to GSH beads and tested for their ability to pulldown WT and mutated PTPRK. These new mutations (illustrated in the new Figure 4 – figure supplement 2) further support the model prediction. F1225A almost completely abolishes binding as predicted, while the M-loop retains binding. These mutations and their effects are now described in the main text and the pull-down data, including controls and retesting of the original DM mutant, are included as panel H in a newly modified Figure 4 focussed solely on the PTPRK interface.
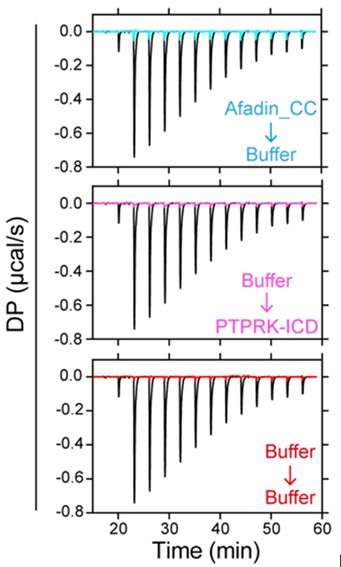
- A minor point is that ITC experiments have not been run long enough to determine the baseline of interaction heats. In addition, as large and polar proteins were used in this experiment, a blank titration would be required to rule out that dilution heats effect the determined affinities.
All control experiments including buffer into buffer, Afadin into buffer and buffer into PTPRK were carried out at the same time as the main binding experiment and are shown below overlaid with the binding curve. These demonstrate the very small dilution heats consistent with excellent buffer matching of the samples.
We were able to obtain excellent fits to the titration curves by fitting 1:1 binding with a calculated linear baseline (see Figure 2B,D). Very similar results were obtained by fitting to the sum (‘composite’) of fitted linear baselines obtained for the three control experiments for each titration.
-
Evaluation Summary:
These studies establish a role for the D2 pseudophosphatase domain of the PTPRK receptor-like phosphotyrosine phosphatase in recruiting Afadin, a cell-cell junction protein that is reported to be a PTPRK substrate, for dephosphorylation by the active D1 phosphatase domain. These findings suggest that the D2 pseudophosphatase domains of RPTPKs might have a general function as platforms to recruit specific phosphotyrosine substrates.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 and Reviewer #3 agreed to share their name with the authors.)
-
Reviewer #1 (Public Review):
The authors have previously reported the identification of a series of cell-cell junctional proteins as pTyr protein targets for the receptor-like PTPRK tyrosine phosphatase (PTP), including Afadin, a junctional plaque protein that links cell surface adhesion proteins to the cytoskeleton. They identified Afadin pY1230 as a target for PTPRK-mediated dephosphorylation, in keeping with the known role of tyrosine phosphorylation in regulating Afadin function in adherens junctions. They also showed that Afadin/PTPRK interaction did not require its tyrosine phosphorylation, and that the whole PTPRK cytoplasmic domain (ICD) was needed for in vitro dephosphorylation of pY1230 Afadin in vitro.
Here, they used two approaches to define a predicted 63-residue coiled-coil (CC) region (residues 1393-1455) in Afadin as …
Reviewer #1 (Public Review):
The authors have previously reported the identification of a series of cell-cell junctional proteins as pTyr protein targets for the receptor-like PTPRK tyrosine phosphatase (PTP), including Afadin, a junctional plaque protein that links cell surface adhesion proteins to the cytoskeleton. They identified Afadin pY1230 as a target for PTPRK-mediated dephosphorylation, in keeping with the known role of tyrosine phosphorylation in regulating Afadin function in adherens junctions. They also showed that Afadin/PTPRK interaction did not require its tyrosine phosphorylation, and that the whole PTPRK cytoplasmic domain (ICD) was needed for in vitro dephosphorylation of pY1230 Afadin in vitro.
Here, they used two approaches to define a predicted 63-residue coiled-coil (CC) region (residues 1393-1455) in Afadin as being sufficient to bind the PTPRK intracellular domain (ICD). However, this region behaved as a monomer suggesting it is not a typical CC region. The CC bound the PTPRK ICD with low μM affinity and interacted selectively with the PTPRK D2 pseudophosphatase domain in vitro. Based on a predicted AlfaFold2/Multimer Afadin CC/D2 domain structure, they biochemically defined the key D2/CC interactions showing that a conserved core charged region, residues 1408-1448, in Afadin was essential, which then allowed them to refine the AlfaFold2 model. Their new model places the Afadin CC core region folded as an α-helix bound across the backside (?) of the D2 domain. They had shown previously that the ICD of the related PTPRU also bound Afadin whereas that of the PTPRM did not, and using the structural model showed that the key contact sites in PTPRK with the Afadin CC helix were conserved in PTPRU but not in PTPRM. When the residues in the G1273/L1335 "acidic" pocket of the D2 domain involved in Afadin helix binding were simultaneously mutated to His and Arg respectively, the basic residues found in PTPRM D2, both the double G1273H/L1335R mutant (DM) D2 alone and the entire PTPRK DM ICD failed to bind Afadin or to dephosphorylate (how much less that WT?) pY1230 in Afadin in lysates of pervanadate-treated cells, as assayed using a pY1230 specific antiserum they generated, even though both the WT and DM PTPRK ICD could dephosphorylate pTyr p120-catenin, another PTPRK substrate. On this basis the authors suggest that the D2 pseudophosphatase domain of PTPRK can act as a substrate recruitment domain that allows the active D1 domain to dephosphorylate a distant pTyr residue, in this case pY1230 ~150 residues away.
In this interesting study, the authors present evidence for the novel concept that the D2 pseudophosphatase domain of PTPRK can serve as a recruitment platform for a subset of PTPRK substrates, such as Afadin. Their evidence for this conclusion is strong, and by extension, their findings suggest that the D2 pseudophosphatase domains of other RPTPs may have a similar general function in substrate recruitment and selectivity.
1. While the AF2-Multimer prediction is quite compelling and supported by the properties of the RPTPK D2 DM mutant, this story would have been even more convincing if they had generated a co-crystal structure (perhaps using a PTPRK D2-Afadin aa 1393-1455 fusion with a long linker). In the absence of a true structure, some additional mutational validation of the proposed Afadin-D2 interaction would strengthen their conclusions.
2. The DM mutant data in Figure 4 show that the D2 domain interaction is important for Afadin pTyr dephosphorylation in vitro, but one would also like evidence that the DM PTPRK mutant lacks Afadin pY1230 dephosphorylating activity in cells. The authors have the PTPRK KO MCF10A cells they generated in their first paper that could be used to re-express the WT and DM PTPRK and then monitor Afadin dephosphorylation with their new anti-pY1230 antibodies.
3. If key residues in PTPRM are mutated into the equivalent PTPRK D2 residues, does this now confer on PTPRM the ability to dephosphorylate pY1230 in Afadin, i.e. a gain of function experiment?
4. It would be helpful to know whether any of the other PTPRK substrates that the authors identified previously have a similar motif that might allow them to bind to the D2 domain and be recruited for dephosphorylation.
-
Reviewer #2 (Public Review):
This study follows up on the authors' previous discovery that afadin is a substrate of PTPRK but not PTPRM and that the D2 domain of PTPRK seems to be necessary for recruitment of afadin. By combining biochemistry on truncation mutations, and modeling the authors demonstrate that the CC domain of afadin can interact with a specific surface of the D2 domain of PTPRK. The authors cleverly leverage similarities and differences between PTPRK vs PTPRM vs PTPRU to identify two residues in PTPRK that are critical for the interaction. Overall the experiments performed are the right ones and the data shown support the conclusions. However it is suggested that the authors provide some more detail about experimental replicates and perform "reverse" mutations of PTPRM to assess whether the residues identified are also …
Reviewer #2 (Public Review):
This study follows up on the authors' previous discovery that afadin is a substrate of PTPRK but not PTPRM and that the D2 domain of PTPRK seems to be necessary for recruitment of afadin. By combining biochemistry on truncation mutations, and modeling the authors demonstrate that the CC domain of afadin can interact with a specific surface of the D2 domain of PTPRK. The authors cleverly leverage similarities and differences between PTPRK vs PTPRM vs PTPRU to identify two residues in PTPRK that are critical for the interaction. Overall the experiments performed are the right ones and the data shown support the conclusions. However it is suggested that the authors provide some more detail about experimental replicates and perform "reverse" mutations of PTPRM to assess whether the residues identified are also sufficient to induce the interaction.
Strengths
This study is of interest to biochemists working on phosphatases especially receptor phosphatases, a group of enzymes whose function and regulation remains poorly understood.Weaknesses
The impact in the field is limited by the fact that afadin is already known to be a D2-dependent substrate of PTPRK. Also this is strictly a biochemistry study and does not provide cellular biology-based evidence in support of the proposed model. -
Reviewer #3 (Public Review):
The study presents interesting new data on the role of the PTPRK D2 pseudophosphatase domain recruiting determining substrate specificity. The paper also demonstrates the utility of predicted structural models, an aspect that has been nicely integrated into this study. However, many open questions remain and additional experimental data should be provided to experimentally confirm the proposed substrate recognition model.
In particular:
- Validation of reagents: The authors generated a pY1230 Afadin antibody claiming that (page 6) "this new antibody is specific to tyrosine phosphorylated Afadin, and that pY1230 is targeted for dephosphorylation by PTPRK, in a D2-domain dependent manner". The WB in Fig 1B shows a lot of background, two main bands are visible which both diminish in intensity in ICT WT …
Reviewer #3 (Public Review):
The study presents interesting new data on the role of the PTPRK D2 pseudophosphatase domain recruiting determining substrate specificity. The paper also demonstrates the utility of predicted structural models, an aspect that has been nicely integrated into this study. However, many open questions remain and additional experimental data should be provided to experimentally confirm the proposed substrate recognition model.
In particular:
- Validation of reagents: The authors generated a pY1230 Afadin antibody claiming that (page 6) "this new antibody is specific to tyrosine phosphorylated Afadin, and that pY1230 is targeted for dephosphorylation by PTPRK, in a D2-domain dependent manner". The WB in Fig 1B shows a lot of background, two main bands are visible which both diminish in intensity in ICT WT pervanadate-treated MCF10A cell lysates. The claim that the developed peptide antibody is selective for pY1230 in Afadin would need to be substantiated, for instance by pull down studies analysed by pY-MS to substantiate a claim of antibody specificity for this site. However, for the current study it would be sufficient to demonstrate that pY1230 is indeed the dephosphorylated site. I suggest therefore including a site directed mutant (Y1230F) that would confirm dephosphorylation at this site and the ability of the pY antibody recognizing the phosphorylation state at this position.
- The authors claim that a short, 63-residue predicted coiled coil (CC) region, is both necessary and sufficient for binding to the PTPRK-ICD. The region is predicted to have alpha-helical structure and as a consequence, a helical structure has been used in the docking model. Considering that the authors recombinantly expressed this region in bacteria, it would be experimentally simple confirming the alpha-helical structure of the segment by CD or NMR spectroscopy.
- Only two mutants have been introduced into PTPRK-ICD to map the Afadin interaction site. One of the mutations changes a possibly structurally important residues (glycine) into a histidine. Even though this residue is present in PTPRM, it does not exclude that the D2 domain no longer functionally folds. Also the second mutation represents a large change in chemical properties and the other 2 predicted residues have not been investigated.
- The interface on the Afadin substrate has not been investigated apart from deleting the entire CC or a central charge cluster. Based on the docking model the authors must have identified key positions of this interaction that could be mutated to confirm the proposed interaction site.
- A minor point is that ITC experiments have not been run long enough to determine the baseline of interaction heats. In addition, as large and polar proteins were used in this experiment, a blank titration would be required to rule out that dilution heats effect the determined affinities.
-
