A translational MRI approach to validate acute axonal damage detection as an early event in multiple sclerosis
Curation statements for this article:-
Curated by eLife

eLife assessment
This paper conducts human and rodent experiments of non-invasive diffusion MRI estimates of axon diameter with the aim to establish whether these estimates provide biologically specific markers of axonal degeneration in MS. It will be of interest to researchers developing quantitative MRI methods and scientists studying neurodegeneration. The experiments provide evidence for the sensitivity of these markers, but do not directly validate axon diameter and do not reflect common pathological mechanisms across rodents and humans.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Axonal degeneration is a central pathological feature of multiple sclerosis and is closely associated with irreversible clinical disability. Current noninvasive methods to detect axonal damage in vivo are limited in their specificity and clinical applicability, and by the lack of proper validation. We aimed to validate an MRI framework based on multicompartment modeling of the diffusion signal (AxCaliber) in rats in the presence of axonal pathology, achieved through injection of a neurotoxin damaging the neuronal terminal of axons. We then applied the same MRI protocol to map axonal integrity in the brain of multiple sclerosis relapsing-remitting patients and age-matched healthy controls. AxCaliber is sensitive to acute axonal damage in rats, as demonstrated by a significant increase in the mean axonal caliber along the targeted tract, which correlated with neurofilament staining. Electron microscopy confirmed that increased mean axonal diameter is associated with acute axonal pathology. In humans with multiple sclerosis, we uncovered a diffuse increase in mean axonal caliber in most areas of the normal-appearing white matter, preferentially affecting patients with short disease duration. Our results demonstrate that MRI-based axonal diameter mapping is a sensitive and specific imaging biomarker that links noninvasive imaging contrasts with the underlying biological substrate, uncovering generalized axonal damage in multiple sclerosis as an early event.
Article activity feed
-

-

Author Response
eLife assesssment:
This paper conducts human and rodent experiments of non-invasive diffusion MRI estimates of axon diameter with the aim to establish whether these estimates provide biologically specific markers of axonal degeneration in MS. It will be of interest to researchers developing quantitative MRI methods and scientists studying neurodegeneration. The experiments provide evidence for the sensitivity of these markers, but do not directly validate axon diameter and do not reflect common pathological mechanisms across rodents and humans.
We thank the Editor for the appreciation of our work. Thanks to the addition of an extensive electron microscopy paradigm, we now include a direct validation of axonal damage and expand on the common pathological mechanisms across the two species. The new results are detailed …
Author Response
eLife assesssment:
This paper conducts human and rodent experiments of non-invasive diffusion MRI estimates of axon diameter with the aim to establish whether these estimates provide biologically specific markers of axonal degeneration in MS. It will be of interest to researchers developing quantitative MRI methods and scientists studying neurodegeneration. The experiments provide evidence for the sensitivity of these markers, but do not directly validate axon diameter and do not reflect common pathological mechanisms across rodents and humans.
We thank the Editor for the appreciation of our work. Thanks to the addition of an extensive electron microscopy paradigm, we now include a direct validation of axonal damage and expand on the common pathological mechanisms across the two species. The new results are detailed in the manuscript and summarized in Fig. 3 in the manuscript
Reviewer #1 (Public Review):
1.1 My primary concern relates to how meaningful the human-rodent comparisons are, and whether these comparisons really advance our understanding of AxCaliber estimates in MS. I applaud the aim to conduct "matched" experiments in both rodent models and human disease. It is a strength that the experiments are aligned with respect to the MRI measurements (although there are some caveats to this mentioned below). But beyond that, the overlap is not what one might hope for: the pathology would seem to be very distinct in humans and rodents, and the histological validation is not specific to what the MRI measurements claim to estimate. To summarize the main findings: (i) in a rat model of general axonal degeneration, axon calibre estimates correlate with neurofilaments; (ii) in MS in humans, axon calibre estimates correlate with demyelinating lesions. This gives a picture of AxCalibre estimates correlating with neuropathology, but is this something that has not already been established in the literature? If the aim is to validate AxCaliber, then there is a logic in using a rodent model that isolates alterations to axonal radius, but what then does this add to the existing literature in that space? If the aim is to study MS (for which AxCaliber results have been previously reported in Huang et al), then why not use a rodent model of MS?
We thank the reviewer for their very insightful comments. Indeed, multiple sclerosis (MS) is a chronic neuroinflammatory and neurodegenerative disease of unknown etiology. An enormous effort has been made to obtain animal models that simulate the pathogenesis of this disease. However, while several models exist recapitulating distinct aspects of the disease (mostly related to demyelination), MS fundamentally remains a disease that only affects humans. This does not mean that EAE or lysolecithin models do not provide information on specific aspects and are therefore valuable. In fact, we believe that trying to replicate the pathological mechanisms of this disease in an animal model goes beyond the scope of the present work. In this work, our intention is to validate a biomarker of axonal damage preclinically, and for this, we use a model of axonal degeneration. We do not claim that this model should be valid to capture the complex clinical and pathological manifestation of MS, but we do think that it is a necessary step to ensure MRI sensitivity to axonal pathology. Why necessary? Because all the available (very limited) MRI literature which provides some form of validation: i) only focuses on healthy tissue, and ii) has an n of 1. Our preclinical paradigm gives conclusive evidence that the MRI axonal diameter proxy detects axonal damage as an increase in the mean diameter. This is now detailed in the discussion.
After this necessary preclinical validation, we then apply the same framework to a human disease like MS that, among other manifestations, is believed to also cause axonal pathology. The improvements with respect to the one published work about axonal diameter in MS are: i) the whole brain analysis, which allowed us to characterize the extent of these early alterations outside the demyelinated lesions; and ii) the larger sample size, which allowed us to uncover an association with disease duration, strengthening our hypothesis about increased axonal diameter being a marker of early disease (new Fig. 5).
Regarding the nonspecificity of histological validation, we thank the reviewer for this insightful comment, which triggered an additional analysis that we believe has added further value to the paper. Using electron microscopy, we found that in our model of neurodegeneration, axonal damage is indeed reflected as an increase in axon diameter (new Fig. 3). These recent findings strongly support the validation of our noninvasive diffusion MRI estimates of axon diameter alterations as an early-stage hallmark of normal-appearing tissue in MS.
Coming back to the comparison between pathology in humans and in rodents, the EM data also support our choice of preclinical model, showing axonal swelling, the same phenomenon reported and characterized in recent postmortem histological data in the normal-appearing white matter of MS patients (Luchicchi et al., Ann Neurol 2021) and in lesions (Fisher et al. Ann Neurol 2007).
All in all, we are confident that the new data supports the validity of this translational approach, and shed new light into the degenerating aspect of MS.
Changes in the manuscript
• Discussion, pag.12: It is important to stress that the aim of this work is not to propose a new animal model of MS, a disease that only affects humans, but rather to validate axonal damage detection (independently from the pathology that has induced it) through noninvasive MRI and apply the framework to characterize axonal pathology in MS.
1.2 I appreciate that both rodent and patient studies are time intensive, major endeavors. Neverthless, the number of subjects is very low in both rodent (n=9) and human (MS=10, control=6) studies. At the very least, this should be more openly acknowledged. But I'm concerned that this is a major weakness of the paper. Related to this, I find it hard to tell how carefully multiple comparison correction was performed throughout. It seems reasonably clear for the TBSS analyses, but then other analyses were performed in ROIs. Are these multiple comparisons corrected as well? Similarly, in Methods, I am confused by the statement that: "post hoc t tests corrected for multiple comparisons whenever a significant effect was detected". What does this mean?
We thank the reviewer for this comment. We agree that a small sample size was a weakness of the previous version of the paper, and therefore, in the new version, we have substantially increased the n for both animal and human experiments (from n=9 to 19 in animals, from 16 to 21 in humans). We removed the ROI analysis in the new version, and thus the confusing statement, and clarified the strategy for multiple comparisons.
Changes in the manuscript
• Data analysis, pag. 18: Lesion masks were excluded from the statistical analysis, and multiple comparisons across clusters were controlled for by using threshold-free cluster enhancement.
1.3 While I do not think the text is in any sense deliberately misleading, I think the authors would do well to either tone down their claims or consider more carefully the implications of the text in many places. Some that stuck out for me are:
Throughout, language in the paper (e.g., "Paired t tests were used to assess differences in the axonal diameter") presumes that the AxCaliber estimates specifically reflect axon diameter. I think the jury is out over whether this is true, particularly for measurements conducted with limited hardware specs. At the very least, I would encourage the author to refer to these measurements throughout as "estimates" of axon diameter.
Thank you for this clarification. We have indeed changed the notation, and now consistently refer to the estimates of axon diameter through MRI as the “MRI axonal diameter proxy”.
1.4 The authors suggest that their results provide "new tools for patient stratification" based on differences in lesion type, but it isn't clear what new information these markers would confer given that the lesions are differentiated based on T1w hypo/hyperintensities. In other words, these lesions are by definition already differentiable from a much simpler MRI marker.
Thank you for this insightful comment. The reviewer is right, and following the general reviewers’ assessment we have decided to not include the lesion analysis in the new version of the manuscript.
1.5 The authors note in the Discussion that: "sensitive to early stages of axonal degeneration, even before alterations in the myelin sheet are detected". Whether intentional or not, the implication in the context of this study is that this would hold for MS (that these markers would detect axonal degeneration preceding demyelination). While there is some discussion of alterations to axonal diameter in MS, the authors do not discuss whether these are the same mechanisms thought to occur in the IBO intervention used here.
Thank you for this comment. Indeed, the scope of the paper is not to assess whether axonal swelling precedes or not myelin alterations, so we agree with the reviewer that this sentence might be misleading and have removed it in the text. While we do not claim that ibotenic acid injections are able to replicate the complex clinical and pathological manifestation of MS (and now we made it clear in the revised manuscript, see comment 1), the electron microscopy paradigm indicates the presence of axonal swelling in the damaged fimbria, which is indeed the same pathological manifestation found in MS post-mortem data (see e.g. Fisher et al. Ann Neurol 2007).
1.6 In the Discussion, the authors note the lack of evidence for a relationship with disability or disease duration, but nevertheless, go on to interpret the "trends" they do observe. I would advise strongly against this: the authors acknowledge that their numbers are low, so I would avoid the temptation to speculate here.
The reviewer is 100% correct. We should have refrained from speculating. In the new version of the paper, however, thanks to the larger human cohort, we were able to find significant associations with disease duration in voxelwise analysis of the white matter skeleton in standard space and in the whole white matter in single subject space (new Figure 5).
1.7 In the Discussion state that "the use of neurofilaments has also been well validated in MS". Well validated for what? MS is a complex disease with a broad range of pathology, so this statement could be read to mean "neurofilaments are known to be altered in MS". However, in the context of this paragraph, the implication would seem to be that neurofilaments are a wellestablished proxy for axonal diameter. Is that the implication, and if so what general evidence is there for this?
We thank the reviewer for this insightful comment. Indeed, altered neurofilaments are not conclusive evidence of increased axonal diameter. In this context, the addition of electron microscopy data in the new manuscript version supports the claim.
Reviewer #2 (Public Review):
Diffusion MRI is sensitive to the brain microstructure, and it has been used to assess the integrity of white matter for nearly 3 decades. Its main limitation is the limited specificity, which makes it difficult to link changes in diffusion parameters to a given pathological substrate. Recently methods based on diffusion MRI that enable the estimation of axonal diameter, non invasively, have become available. This paper aims at validating one of such methods using an experimental model of neurodegeneration. The authors found a significant correlation between axonal diameter estimated by MRI and an histological marker of neurodegeneration. Although this is of great interest, as it demonstrates that this method is sensitive to neurodegeneration, a direct validation would require a measurement of axonal diameter using electron or confocal microscopy, rather than a correlation with a measure of axonal degeneration not directly related to axonal diameter. So, although these data are compelling, they do not prove that the increase in axonal diameter suggested by diffusion MRI corresponds to actual axonal swelling. The Authors also apply the same method to compare the white matter of patients with multiple sclerosis (MS) and healthy controls, showing widespread increases in axonal diameter in the patients. These data are compelling, but again, not conclusive. Other factors such as gloss could bias the MRI measurement and lead to an apparent increase in axonal diameter.
We would like to thank the reviewer for the positive assessment of our work and for the valuable suggestion. We are confident that the new version of the manuscript, by including an extensive validation based on electron microscopy, has addressed the reviewer´s criticisms.
Reviewer #3 (Public Review):
3.1 In this paper, Toschi et al. performed dMRI to in vivo estimate axon diameter in the brain and demonstrated that multi-compartmental modeling (AxCaliber) is sensitive to microstructural axonal damage in rats and axon caliber increase in demyelinating lesions in MS patients, suggesting that axon diameter mapping provides a potential biomarker to bridge the gap between medical imaging contrasts and biological microstructure. In particular, authors injected ibotenic acid (IBO) and saline in the left and right rat hippocampus, respectively, and compared in vivo estimated axon diameter and ex vivo neurofilament staining in left and right fimbria. The axon size estimation was larger in the fimbria of IBO injection side, where the neurofilament intensity is higher. Correlation of axon size estimation and neurofilament intensity was observed in both injection sides. Further, higher axon diameter estimation was observed in normal appearing white matter (NAWM) of MS patients, compared with the healthy subjects. The axon size estimation increased in hypointense lesions of T1 weighted contrast, but not in isointense lesions. Through the comparison of dMRI-estimated axon size and histology-based fluorescence intensity, authors indirectly validated the sensitivity of axon diameter mapping to the tissue microstructure in the rat brain, and further explored the axon size change in the brain of MS patients. However, the dMRI protocol and biophysical modeling in this study were not fully optimized to maximize the sensitivity to axon size estimation, and the dMRI-estimated axon size (4.4-5.4 micron) was much larger than values reported in previous histological studies (0.5-3 micron) [Barazany et al., Brain 2009]. Finally, although the modified AxCaliber model incorporated two fiber bundles in different directions, the fiber dispersion in each bundle was not considered (c.f. fiber dispersion ~20-30 degree in corpus callosum), potentially leading to overestimated axon diameter.
We thank the reviewer for their appreciation of our work, which we believe is substantially improved in this revised version through the inclusion of an electron microscopy paradigm. Below, the point-by-point response to the specific points raised.
3.2 The conclusions in this study are supported by experimental results. However, the dMRI protocol and biophysical model could be further optimized and validated: 1. To in vivo estimate the axon diameter ~1 micron using dMRI, strong diffusion weighting (b-value) should be applied to maximize the signal decay due to intra-axonal restricted diffusion and minimize the signal contribution of extra-cellular hindered diffusion. However, authors only applied maximal b-value = 4000 s/mm2, much smaller than values ~15,00020,000 s/mm2 in previous studies [Assaf et al., MRM 2008; Huang et al., BSAF 2020, 225:1277]. The use of low diffusion weighting in this study leads to a lower bound ~4-6 micron for accurate diameter estimation, the so-called resolution limit in [Nilsson et al., NMR Biomed 2017, 30:e3711]. In other words, the estimated axon diameter is potentially overestimated and related with the imaging protocol and image quality, confounding the biological interpretation.
We thank the reviewer for this insightful comment. Indeed, while the resolution limit is a concern, the chosen b-value has been a compromise between sensitivity to small structure and SNR, as indicated by recent animal (Crater et al., 2022) and human (Jensen et al., 2016; McKinnon et al., 2017; Moss et al., 2019) work, pointing at 3000-4000 s/mm2 as the b-value for which the intra-axonal water signal is dominant. In addition, a paper from the laboratory that first developed the Axcaliber method recently came out (Gast et al., 2023, DOI: 10.1007/s12021-023-09630-w) demonstrating that an MRI protocol with a maximum b-value between 3000 and 4000 s/mm2 (and even lower) is sufficient to capture, in vivo and in humans, various well-known aspects of axonal morphometry (e.g., the corpus callosum axon diameter variation) as well as other aspects that are less explored (e.g., axon diameter-based separation of the superior longitudinal fasciculus into segments). The same paper contains resources and further bibliography supporting the fact that experimental evidence suggests that the contribution of intra-axonal water to restricted diffusion signals dominates other factors (see Online Resource 1, section A of the same paper). To challenge this recent evidence from a neurobiology perspective, we include in the supplementary material a subset of experiments in animals with lower maximum b-value (2500 s/mm2, Fig. S1), where we are able to detect the same effect of increased MRI axonal diameter proxy in the injected hemisphere compared to control.
We would like to add that while extremely valuable and informative, simulation studies such as the excellent study by Veraart et al., 2020, are inevitably valid under certain assumptions. Among them, some critical ones are i) the need to neglect nonaxonal cells such as glia, ii) assuming that the bulk diffusivity of water in cerebral tissue would be the same as that of free water, and iii) impermeable barriers. All these assumptions are expected to play a role in the estimated resolution limit, a role difficult to quantify but likely substantial.
For this reason, we believe that our approach, which is 100% focused on neurobiology and measurements performed in real tissue, can offer a different perspective and fuel the ongoing debate on axonal diameter measurement feasibility. We acknowledge the value of the reviewer comment and discuss the issue of b-value in the discussion (see also comment 1.8).
Changes in the manuscript
• Discussion, pag. 12:
Despite some inevitable minor differences due to different brain sizes and magnet features, the human protocol was built to match the main characteristics of the preclinical diffusion sequence, such as the b-value and diffusion time range. The chosen b-value has been a compromise between sensitivity to small structures and the signalto-noise ratio (SNR), as indicated by recent animal (Crater et al., 2022) and human (Gast et al., 2023; Jensen et al., 2016; McKinnon et al., 2017; Moss et al., 2019) work, pointing at 4000 s/mm2 as the b-value for which the intra-axonal water signal is dominant. However, following recent work supporting sensitivity of diffusion-weighted MRI to axonal diameter even at lower b-values (Gast et al., 2023), we tested a protocol with a lower b-value in a subset of animals, with the aim of facilitating future clinical AxCaliber studies. We found no qualitative differences in the outcome (MRI axonal diameter proxy was increased following fimbria damage). Further work and perhaps more realistic simulations, considering real cell composition and morphology, are needed to clarify this issue.3.3 In this study, the positive correlation of dMRI-estimated axon size and neurofilament fluorescence intensity is indeed an encouraging result, and yet this validation is indirect since it relies on the positive correlation between neurofilament intensity and axon diameter in histology.
The reviewer correctly points out a severe limitation of the previous manuscript version, which is now addressed by including an extensive electron microscopy evaluation, recapitulated in new Fig. 3.
3.4 Authors did not consider the fiber dispersion in the proposed dMRI model. This can lead to overestimated axon diameter, even in the highly aligned WM, such as corpus callosum with ~20-30 degree dispersion in histology [Ronen et al., BSAF 2014, 219:1773; Leergaard et all, PLoS One 2010, 5(1), e8595] and MRI [Dhital et al., NeuroImage 2019, 189, 543; Novikov et al., NeuroImage 2018, 174:518].
The reviewer is correctly pointing out an important characteristic of while matter microstructure as is fibre dispersion. However, we would like to point out that the use of a second fiber population is expected to mitigate this effect by absorbing some axonal directional dispersion in areas of a single fiber. To support this, we quantified dispersion as the angle between the two main fiber orientations captured by the AxCaliber fit, as showed in Author response image 1 for two representative subjects (one control, upper line, and one MS, lower line; the “dispersion” maps are masked by a white matter probability mask, and superimposed to a T2w). Indeed, the angle between the two main fibres in the corpus callosum is around 20 degrees or lower, compatible with the bibliography cited by the reviewer, and higher in other white matter areas known to be characterized by fiber crossing and dispersion.
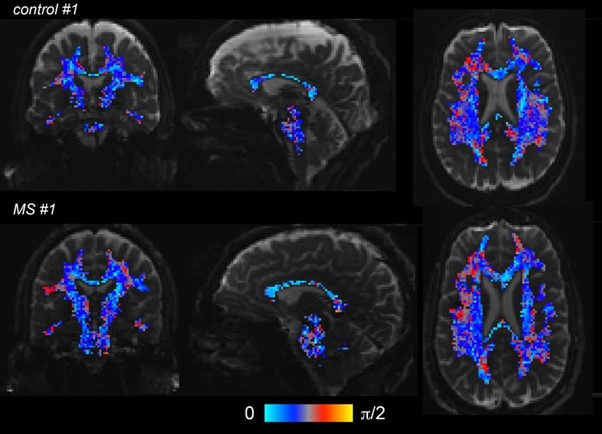
Author response image 1.
Angle in radians between the two main fiber orientations captured by the AxCaliber fit, as showed below for two representative subjects (one control, upper line, and one MS, lower line). The dispersion maps are masked by a white matter probability mask (P>=0.95), and superimposed to a T2-weighted image.
-
eLife assessment
This paper conducts human and rodent experiments of non-invasive diffusion MRI estimates of axon diameter with the aim to establish whether these estimates provide biologically specific markers of axonal degeneration in MS. It will be of interest to researchers developing quantitative MRI methods and scientists studying neurodegeneration. The experiments provide evidence for the sensitivity of these markers, but do not directly validate axon diameter and do not reflect common pathological mechanisms across rodents and humans.
-
Reviewer #1 (Public Review):
1. My primary concern relates to how meaningful the human-rodent comparisons are, and whether these comparisons really advance our understanding of AxCaliber estimates in MS.
I applaud the aim to conduct "matched" experiments in both rodent models and human disease. It is a strength that the experiments are aligned with respect to the MRI measurements (although there are some caveats to this mentioned below). But beyond that, the overlap is not what one might hope for: the pathology would seem to be very distinct in humans and rodents, and the histological validation is not specific to what the MRI measurements claim to estimate.
To summarize the main findings: (i) in a rat model of general axonal degeneration, axon calibre estimates correlate with neurofilaments; (ii) in MS in humans, axon calibre estimates …
Reviewer #1 (Public Review):
1. My primary concern relates to how meaningful the human-rodent comparisons are, and whether these comparisons really advance our understanding of AxCaliber estimates in MS.
I applaud the aim to conduct "matched" experiments in both rodent models and human disease. It is a strength that the experiments are aligned with respect to the MRI measurements (although there are some caveats to this mentioned below). But beyond that, the overlap is not what one might hope for: the pathology would seem to be very distinct in humans and rodents, and the histological validation is not specific to what the MRI measurements claim to estimate.
To summarize the main findings: (i) in a rat model of general axonal degeneration, axon calibre estimates correlate with neurofilaments; (ii) in MS in humans, axon calibre estimates correlate with demyelinating lesions. This gives a picture of AxCalibre estimates correlating with neuropathology, but is this something that has not already been established in the literature?
If the aim is to validate AxCaliber, then there is a logic in using a rodent model that isolates alterations to axonal radius, but what then does this add to the existing literature in that space? If the aim is to study MS (for which AxCaliber results have been previously reported in Huang et al), then why not use a rodent model of MS?
2. I appreciate that both rodent and patient studies are time intensive, major endeavors. Neverthless, the number of subjects is very low in both rodent (n=9) and human (MS=10, control=6) studies. At the very least, this should be more openly acknowledged. But I'm concerned that this is a major weakness of the paper. Related to this, I find it hard to tell how carefully multiple comparison correction was performed throughout. It seems reasonably clear for the TBSS analyses, but then other analyses were performed in ROIs. Are these multiple comparisons corrected as well? Similarly, in Methods, I am confused by the statement that: "post hoc t tests corrected for multiple comparisons whenever a significant effect was detected". What does this mean?
3. While I do not think the text is in any sense deliberately misleading, I think the authors would do well to either tone down their claims or consider more carefully the implications of the text in many places. Some that stuck out for me are:
(a) Throughout, language in the paper (e.g., "Paired t tests were used to assess differences in the axonal diameter") presumes that the AxCaliber estimates specifically reflect axon diameter. I think the jury is out over whether this is true, particularly for measurements conducted with limited hardware specs. At the very least, I would encourage the author to refer to these measurements throughout as "estimates" of axon diameter.
(b) The authors suggest that their results provide "new tools for patient stratification" based on differences in lesion type, but it isn't clear what new information these markers would confer given that the lesions are differentiated based on T1w hypo/hyperintensities. In other words, these lesions are by definition already differentiable from a much simpler MRI marker.
(c) The authors note in the Discussion that: "sensitive to early stages of axonal degeneration, even before alterations in the myelin sheet are detected". Whether intentional or not, the implication in the context of this study is that this would hold for MS (that these markers would detect axonal degeneration preceding demyelination). While there is some discussion of alterations to axonal diameter in MS, the authors do not discuss whether these are the same mechanisms thought to occur in the IBO intervention used here.
(d) In the Discussion, the authors note the lack of evidence for a relationship with disability or disease duration, but nevertheless, go on to interpret the "trends" they do observe. I would advise strongly against this: the authors acknowledge that their numbers are low, so I would avoid the temptation to speculate here.
(e) In the Discussion state that "the use of neurofilaments has also been well validated in MS". Well validated for what? MS is a complex disease with a broad range of pathology, so this statement could be read to mean "neurofilaments are known to be altered in MS". However, in the context of this paragraph, the implication would seem to be that neurofilaments are a well-established proxy for axonal diameter. Is that the implication, and if so what general evidence is there for this?
-
Reviewer #2 (Public Review):
Diffusion MRI is sensitive to the brain microstructure, and it has been used to assess the integrity of white matter for nearly 3 decades. Its main limitation is the limited specificity, which makes it difficult to link changes in diffusion parameters to a given pathological substrate. Recently methods based on diffusion MRI that enable the estimation of axonal diameter, non invasively, have become available. This paper aims at validating one of such methods using an experimental model of neurodegeneration. The authors found a significant correlation between axonal diameter estimated by MRI and an histological marker of neurodegeneration. Although this is of great interest, as it demonstrates that this method is sensitive to neurodegeneration, a direct validation would require a measurement of axonal …
Reviewer #2 (Public Review):
Diffusion MRI is sensitive to the brain microstructure, and it has been used to assess the integrity of white matter for nearly 3 decades. Its main limitation is the limited specificity, which makes it difficult to link changes in diffusion parameters to a given pathological substrate. Recently methods based on diffusion MRI that enable the estimation of axonal diameter, non invasively, have become available. This paper aims at validating one of such methods using an experimental model of neurodegeneration. The authors found a significant correlation between axonal diameter estimated by MRI and an histological marker of neurodegeneration. Although this is of great interest, as it demonstrates that this method is sensitive to neurodegeneration, a direct validation would require a measurement of axonal diameter using electron or confocal microscopy, rather than a correlation with a measure of axonal degeneration not directly related to axonal diameter. So, although these data are compelling, they do not prove that the increase in axonal diameter suggested by diffusion MRI corresponds to actual axonal swelling. The Authors also apply the same method to compare the white matter of patients with multiple sclerosis (MS) and healthy controls, showing widespread increases in axonal diameter in the patients. These data are compelling, but again, not conclusive. Other factors such as gloss could bias the MRI measurement and lead to an apparent increase in axonal diameter.
-
Reviewer #3 (Public Review):
In this paper, Toschi et al. performed dMRI to in vivo estimate axon diameter in the brain and demonstrated that multi-compartmental modeling (AxCaliber) is sensitive to microstructural axonal damage in rats and axon caliber increase in demyelinating lesions in MS patients, suggesting that axon diameter mapping provides a potential biomarker to bridge the gap between medical imaging contrasts and biological microstructure. In particular, authors injected ibotenic acid (IBO) and saline in the left and right rat hippocampus, respectively, and compared in vivo estimated axon diameter and ex vivo neurofilament staining in left and right fimbria. The axon size estimation was larger in the fimbria of IBO injection side, where the neurofilament intensity is higher. Correlation of axon size estimation and …
Reviewer #3 (Public Review):
In this paper, Toschi et al. performed dMRI to in vivo estimate axon diameter in the brain and demonstrated that multi-compartmental modeling (AxCaliber) is sensitive to microstructural axonal damage in rats and axon caliber increase in demyelinating lesions in MS patients, suggesting that axon diameter mapping provides a potential biomarker to bridge the gap between medical imaging contrasts and biological microstructure. In particular, authors injected ibotenic acid (IBO) and saline in the left and right rat hippocampus, respectively, and compared in vivo estimated axon diameter and ex vivo neurofilament staining in left and right fimbria. The axon size estimation was larger in the fimbria of IBO injection side, where the neurofilament intensity is higher. Correlation of axon size estimation and neurofilament intensity was observed in both injection sides. Further, higher axon diameter estimation was observed in normal appearing white matter (NAWM) of MS patients, compared with the healthy subjects. The axon size estimation increased in hypointense lesions of T1 weighted contrast, but not in isointense lesions. Through the comparison of dMRI-estimated axon size and histology-based fluorescence intensity, authors indirectly validated the sensitivity of axon diameter mapping to the tissue microstructure in the rat brain, and further explored the axon size change in the brain of MS patients. However, the dMRI protocol and biophysical modeling in this study were not fully optimized to maximize the sensitivity to axon size estimation, and the dMRI-estimated axon size (4.4-5.4 micron) was much larger than values reported in previous histological studies (0.5-3 micron) [Barazany et al., Brain 2009]. Finally, although the modified AxCaliber model incorporated two fiber bundles in different directions, the fiber dispersion in each bundle was not considered (c.f. fiber dispersion ~20-30 degree in corpus callosum), potentially leading to overestimated axon diameter.
The conclusions in this study are supported by experimental results. However, the dMRI protocol and biophysical model could be further optimized and validated:
1. To in vivo estimate the axon diameter ~1 micron using dMRI, strong diffusion weighting (b-value) should be applied to maximize the signal decay due to intra-axonal restricted diffusion and minimize the signal contribution of extra-cellular hindered diffusion. However, authors only applied maximal b-value = 4000 s/mm2, much smaller than values ~15,000-20,000 s/mm2 in previous studies [Assaf et al., MRM 2008; Huang et al., BSAF 2020, 225:1277]. The use of low diffusion weighting in this study leads to a lower bound ~4-6 micron for accurate diameter estimation, the so-called resolution limit in [Nilsson et al., NMR Biomed 2017, 30:e3711]. In other words, the estimated axon diameter is potentially overestimated and related with the imaging protocol and image quality, confounding the biological interpretation.
2. In this study, the positive correlation of dMRI-estimated axon size and neurofilament fluorescence intensity is indeed an encouraging result, and yet this validation is indirect since it relies on the positive correlation between neurofilament intensity and axon diameter in histology.
3. Authors did not consider the fiber dispersion in the proposed dMRI model. This can lead to overestimated axon diameter, even in the highly aligned WM, such as corpus callosum with ~20-30 degree dispersion in histology [Ronen et al., BSAF 2014, 219:1773; Leergaard et all, PLoS One 2010, 5(1), e8595] and MRI [Dhital et al., NeuroImage 2019, 189, 543; Novikov et al., NeuroImage 2018, 174:518]. -
