Genome-wide base editor screen identifies regulators of protein abundance in yeast
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
Schubert and coworkers report the development of a novel CRISPR-based screening method that allows probing interactions between (a large set of) specific mutations and the abundance of specific proteins, and, more generally, investigate the spectrum of effects that (point) mutations can have on protein abundance. This complements existing strategies for measuring effects of genetic perturbations on transcript levels, which is important as for some proteins mRNA and protein levels do not correlate well. The ability to measure proteins directly therefore promises to close an important gap in our understanding of the links between genotype and phenotype, and the strategy is broadly applicable beyond the current study.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 and Reviewer #2 agreed to share their name with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Proteins are key molecular players in a cell, and their abundance is extensively regulated not just at the level of gene expression but also post-transcriptionally. Here, we describe a genetic screen in yeast that enables systematic characterization of how protein abundance regulation is encoded in the genome. The screen combines a CRISPR/Cas9 base editor to introduce point mutations with fluorescent tagging of endogenous proteins to facilitate a flow-cytometric readout. We first benchmarked base editor performance in yeast with individual gRNAs as well as in positive and negative selection screens. We then examined the effects of 16,452 genetic perturbations on the abundance of eleven proteins representing a variety of cellular functions. We uncovered hundreds of regulatory relationships, including a novel link between the GAPDH isoenzymes Tdh1/2/3 and the Ras/PKA pathway. Many of the identified regulators are specific to one of the eleven proteins, but we also found genes that, upon perturbation, affected the abundance of most of the tested proteins. While the more specific regulators usually act transcriptionally, broad regulators often have roles in protein translation. Overall, our novel screening approach provides unprecedented insights into the components, scale and connectedness of the protein regulatory network.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
Overall, the science is sound and interesting, and the results are clearly presented. However, the paper falls in-between describing a novel method and studying biology. As a consequence, it is a bit difficult to grasp the general flow, central story and focus point. The study does uncover several interesting phenomena, but none are really studied in much detail and the novel biological insight is therefore a bit limited and lost in the abundance of observations. Several interesting novel interactions are uncovered, in particular for the SPS sensor and GAPDH paralogs, but these are not followed up on in much detail. The same can be said for the more general observations, eg the fact that different types of mutations (missense vs nonsense) in different types of genes (essential vs …
Author Response
Reviewer #1 (Public Review):
Overall, the science is sound and interesting, and the results are clearly presented. However, the paper falls in-between describing a novel method and studying biology. As a consequence, it is a bit difficult to grasp the general flow, central story and focus point. The study does uncover several interesting phenomena, but none are really studied in much detail and the novel biological insight is therefore a bit limited and lost in the abundance of observations. Several interesting novel interactions are uncovered, in particular for the SPS sensor and GAPDH paralogs, but these are not followed up on in much detail. The same can be said for the more general observations, eg the fact that different types of mutations (missense vs nonsense) in different types of genes (essential vs non-essential, housekeeping vs. stress-regulated...) cause different effects.
This is not to say that the paper has no merit - far from it even. But, in its current form, it is a bit chaotic. Maybe there is simply too much in the paper? To me, it would already help if the authors would explicitly state that the paper is a "methods" paper that describes a novel technique for studying the effects of mutations on protein abundance, and then goes on to demonstrate the possibilities of the technology by giving a few examples of the phenomena that can be studied. The discussion section ends in this way, but it may be helpful if this was moved to the end of the introduction.
We modified the manuscript as suggested.
Reviewer #2 (Public Review):
Schubert et al. describe a new pooled screening strategy that combines protein abundance measurements of 11 proteins determined via FACS with genome-wide mutagenesis of stop codons and missense mutations (achieved via a base editor) in yeast. The method allows to identify genetic perturbations that affect steady state protein levels (vs transcript abundance), and in this way define regulators of protein abundance. The authors find that perturbation of essential genes more often alters protein abundance than of nonessential genes and proteins with core cellular functions more often decrease in abundance in response to genetic perturbations than stress proteins. Genes whose knockouts affected the level of several of the 11 proteins were enriched in protein biosynthetic processes while genes whose knockouts affected specific proteins were enriched for functions in transcriptional regulation. The authors also leverage the dataset to confirm known and identify new regulatory relationships, such as a link between the SDS amino acid sensor and the stress response gene Yhb1 or between Ras/PKA signalling and GAPDH isoenzymes Tdh1, 2, and 3. In addition, the paper contains a section on benchmarking of the base editor in yeast, where it has not been used before.
Strengths and weaknesses of the paper
The authors establish the BE3 base editor as a screening tool in S. cerevisiae and very thoroughly benchmark its functionality for single edits and in different screening formats (fitness and FACS screening). This will be very beneficial for the yeast community.
The strategy established here allows measuring the effect of genetic perturbations on protein abundances in highly complex libraries. This complements capabilities for measuring effects of genetic perturbations on transcript levels, which is important as for some proteins mRNA and protein levels do not correlate well. The ability to measure proteins directly therefore promises to close an important gap in determining all their regulatory inputs. The strategy is furthermore broadly applicable beyond the current study. All experimental procedures are very well described and plasmids and scripts are openly shared, maximizing utility for the community.
There is a good balance between global analyses aimed at characterizing properties of the regulatory network and more detailed analyses of interesting new regulatory relationships. Some of the key conclusions are further supported by additional experimental evidence, which includes re-making specific mutations and confirming their effects on protein levels by mass spectrometry.
The conclusions of the paper are mostly well supported, but I am missing some analyses on reproducibility and potential confounders and some of the data analysis steps should be clarified.
The paper starts on the premise that measuring protein levels will identify regulators and regulatory principles that would not be found by measuring transcripts, but since the findings are not discussed in light of studies looking at mRNA levels it is unclear how the current study extends knowledge regarding the regulatory inputs of each protein.
See response to Comment #10.
Specific comments regarding data analysis, reproducibility, confounders
- The authors use the number of unique barcodes per guide RNA rather than barcode counts to determine fold-changes. For reliable fold changes the number of unique barcodes per gRNA should then ideally be in the 100s for each guide, is that the case? It would also be important to show the distribution of the number of barcodes per gRNA and their abundances determined from read counts. I could imagine that if the distribution of barcodes per gRNA or the abundance of these barcodes is highly skewed (particularly if there are many barcodes with only few reads) that could lead to spurious differences in unique barcode number between the high and low fluorescence pool. I imagine some skew is present as is normal in pooled library experiments. The fold-changes in the control pools could show whether spurious differences are a problem, but it is not clear to me if and how these controls are used in the protein screen.
Because of the large number of screens performed in this study (11 proteins, with 8 replicates for each) we had to trade off sequencing depth and power against cell sorting time and sequencing cost, resulting in lower read and barcode numbers than what might be ideally aimed for. As described further in the response to Comment #5, we added a new figure to the manuscript that shows that the correlation of fold-changes between replicates is high (Figure 3–S1A). The second figure below shows that the correlation between the number of unique barcodes and the number of reads per gRNA is highly significant (p < 2.2e-16).
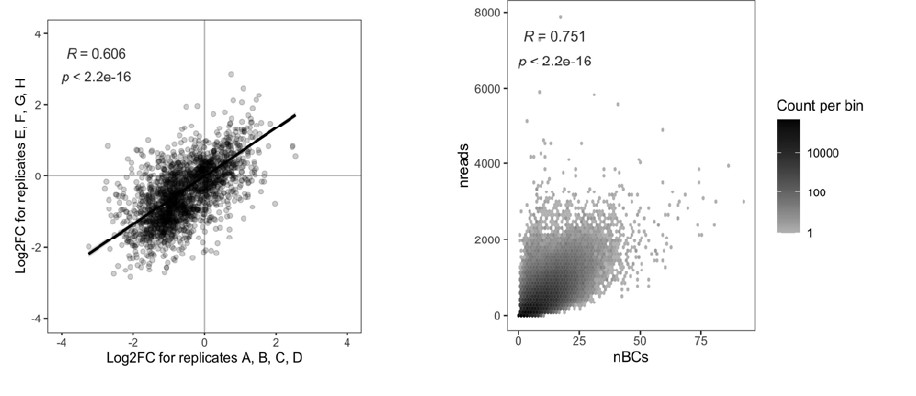
- I like the idea of using an additional barcode (plasmid barcode) to distinguish between different cells with the same gRNA - this would directly allow to assess variability and serve as a sort of replicate within replicate. However, this information is not leveraged in the analysis. It would be nice to see an analysis of how well the different plasmid barcodes tagging the same gRNA agree (for fitness and protein abundance), to show how reproducible and reliable the findings are.
We agree with the reviewer that this would be nice to do in principle, but our sequencing depth for the sorted cell populations was not high enough to compare the same barcode across the low/unsorted/high samples. See also our response to Comment #5 for the replicate analyses.
- From Fig 1 and previous research on base editors it is clear that mutation outcomes are often heterogeneous for the same gRNA and comprise a substantial fraction of wild-type alleles, alleles where only part of the Cs in the target window or where Cs outside the target window are edited, and non C-to-T edits. How does this reflect on the variability of phenotypic measurements, given that any barcode represents a genetically heterogeneous population of cells rather than a specific genotype? This would be important information for anyone planning to use the base editor in future.
We agree with the reviewer that the heterogeneity of editing outcomes is an important point to keep in mind when working with base editors. In genetic screens, like the ones described here, often the individual edit is less important, and the overall effects of the base editor are specific/localized enough to obtain insights into the effects of mutations in the area where the gRNA targets the genome. For example, in our test screens for Canavanine resistance and fitness effects, in which we used gRNAs predicted to introduce stop codons into the CAN1 gene and into essential genes, respectively, we see the expected loss-of-function effect for a majority of the gRNAs (canavanine screen: expected effect for 67% of all gRNAs introducing stop codons into CAN1; fitness screen: expected effect for 59% of all gRNAs introducing stop codons into essential genes) (Figure 2). In the canavanine screen, we also see that gRNAs predicted to introduce missense mutations at highly conserved residues are more likely to lead to a loss-of-function effect than gRNAs predicted to introduce missense mutations at less conserved residues, further highlighting the differentiated results that can be obtained with the base editor despite the heterogeneity in editing outcomes overall. We would certainly advise anyone to confirm by sequencing the base edits in individual mutants whenever a precise mutation is desired, as we did in this study when following up on selected findings with individual mutants.
- How common are additional mutations in the genome of these cells and could they confound the measured effects? I can think of several sources of additional mutations, such as off-target editing, edits outside the target window, or when 2 gRNA plasmids are present in the same cell (both target windows obtain edits). Could some of these events explain the discrepancy in phenotype for two gRNAs that should make the same mutation (Fig S4)? Even though BE3 has been described in mammalian cells, an off-target analysis would be desirable as there can be substantial differences in off-target behavior between cell types and organisms.
Generally, we are not very concerned about random off-target activity of the base editor because we would not expect this to cause a consistent signal that would be picked up in our screen as a significant effect of a particular gRNA. Reproducible off-target editing with a specific gRNA at a site other than the intended target site would be problematic, though. We limited the chance of this happening by not using gRNAs that may target similar sequences to the intended target site in the genome. Specifically, we excluded gRNAs that have more than one target in the genome when the 12 nucleotides in the seed region (directly upstream of the PAM site) are considered (DiCarlo et al., Nucleic Acids Research, 2013).
We do observe some off-target editing right outside the target window, but generally at much lower frequency than the on-target editing in the target window (Figure 1B and Figure 1–S2). Since for most of our analyses we grouped perturbations per gene, such off-target edits should not affect our findings. In addition, we validated key findings with independent experiments. For our study, we used the Base Editor v3 (Komor et al., Nature, 2016); more recently, additional base editors have been developed that show improved accuracy and efficiency, and we would recommend these base editors when starting a new study (see, e.g., Anzalone et al., Nature Biotechnology, 2020).
We are not concerned about cases in which one cell gets two gRNAs, since the chance that the same two gRNAs end up in one cell repeatedly is low, and such events would therefore not result in a significant signal in our screens.
We don’t think that off-target mutations can explain the discrepancy between pairs of gRNAs that should introduce the same mutation (Figure 3–S1. The effect of the two gRNAs is actually well-correlated, but, often, one of the two gRNAs doesn’t pass our significance cut-off or simply doesn’t edit efficiently (i.e., most discrepancies arise from false negatives rather than false positives). We may therefore miss the effects of some mutations, but we are unlikely to draw erroneous conclusions from significant signals.
- In the protein screen normalization uses the total unique barcode counts. Does this efficiently correct for differences from sequencing (rather than total read counts or other methods)? It would be nice to see some replicate plots for the analysis of the fitness as well as the protein screen to be able to judge that.
We made a new figure that shows a replicate comparison for the protein screen (see below; in the manuscript it is Figure 3–S1A) and commented on it in the manuscript. For this analysis, the eight replicates for each protein were split into two groups of four replicates each and analyzed the same way as the eight replicates. The correlation between the two groups of replicates is highly significant (p < 2.2e-16). The second figure shows that the total number of reads and the total number of unique barcodes are well correlated.
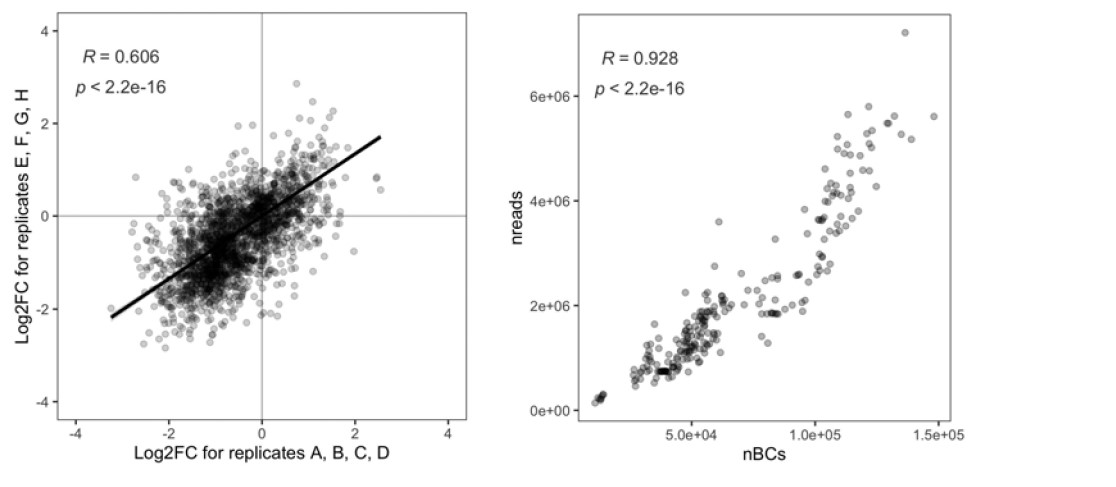
For the fitness screen, we used read counts rather than barcode counts for the analysis since read counts better reflect the dropout of cells due to reduced fitness. The figure below shows a replicate comparison for the fitness screen. For this analysis, the four replicates were split into two groups of two replicates each and analyzed the same way as the four replicates. The correlation between the two groups of replicates is highly significant (p < 2.2e-16).
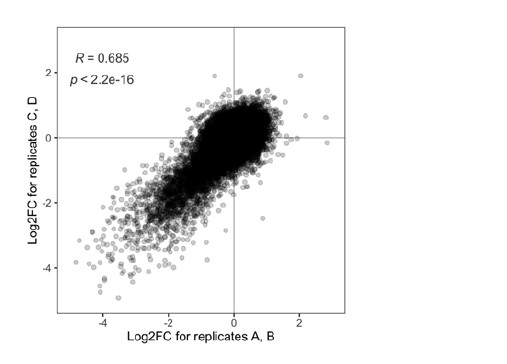
- In the main text the authors mention very high agreement between gRNAs introducing the same mutation but this is only based on 20 or so gRNA pairs; for many more pairs that introduce the same mutation only one reaches significance, and the correlation in their effects is lower (Fig S4). It would be better to reflect this in the text directly rather than exclusively in the supplementary information.
We clarified this in the manuscript main text: “For 78 of these gRNA pairs, at least one gRNA had a significant effect (FDR < 0.05) on at least one of the eleven proteins; their effects were highly correlated (Pearson’s R2 = 0.43, p < 2.2E-16) (Figure 3–S1B). For the 20 gRNA pairs for which both gRNAs had a significant effect, the correlation was even higher (Pearson’s R2 = 0.819, p = 8.8e-13) (Figure 3–S1C). These findings show that the significant gRNA effects that we identify have a low false positive rate, but they also suggest that many real gRNA effects are not detected in the screen due to limitations in statistical power.”
- When the different gRNAs for a targeted gene are combined, instead of using an averaged measure of their effects the authors use the largest fold-change. This seems not ideal to me as it is sensitive to outliers (experimental error or background mutations present in that strain).
We agree that the method we used is more sensitive to outliers than averaging per gene. However, because many gRNAs have no effect either because they are not editing efficiently or because the edit doesn’t have a phenotypic consequence, an averaging method across all gRNAs targeting the same gene would be too conservative and not properly capture the effect of a perturbation of that gene.
- Phenotyping is performed directly after editing, when the base editor is still present in the cells and could still interact with target sites. I could imagine this could lead to reduced levels of the proteins targeted for mutagenesis as it could act like a CRISPRi transcriptional roadblock. Could this enhance some of the effects or alter them in case of some missense mutations?
To reduce potential “CRISPRi-like” effects of the base editor on gene expression, we placed the base editor under a galactose-inducible promoter. For both the fitness and protein screens we grew the cultures in media without galactose for another 24 hours (fitness screen) or 8-9 hours (protein screens) before sampling. In the latter case, this recovery time corresponded to more than three cell divisions, after which we assume base editor levels to have strongly decreased, and therefore to no longer interfere with transcription. This is also supported by our ability to detect discordant effects of gRNAs targeting the same gene (e.g., the two mutations leading to loss-of-function and gain-of-function of RAS2), which would otherwise be overshadowed by a CRISPRi effect.
- I feel that the main text does not reflect the actual editing efficiency very well (the main numbers I noticed were 95% C to T conversion and 89% of these occurring in a specific window). More informative for interpreting the results would be to know what fraction of the alleles show an edit (vs wild-type) and how many show the 'complete' edit (as the authors assume 100% of the genotypes generated by a gRNA to be conversion of all Cs to Ts in the target window). It would be important to state in the main text how variable this is for different gRNAs and what the typical purity of editing outcomes is.
We now show the editing efficiency and purity in a new figure (Figure 1B), and discuss it in the main text as follows: “We found that the target window and mutagenesis pattern are very similar to those described in human cells: 95% of edits are C-to-T transitions, and 89% of these occurred in a five-nucleotide window 13 to 17 base pairs upstream of the PAM sequence (Figure 1A; Figure 1–S2) (Komor et al., 2016). Editing efficiency was variable across the eight gRNAs and ranged from 4% to 64% if considering only cases where all Cs in the window are edited; percentages are higher if incomplete edits are considered, too (Figure 1B).”
Comments regarding findings
- It would be nice to see a comparison of the results to the effects of ~1500 yeast gene knockouts on cellular transcriptomes (https://doi.org/10.1016/j.cell.2014.02.054). This would show where the current study extends established knowledge regarding the regulatory inputs of each protein and highlight the importance of directly measuring protein levels. This would be particularly interesting for proteins whose abundance cannot be predicted well from mRNA abundance.
We agree with the reviewer that it would be very interesting to compare the effect of perturbations on mRNA vs protein levels. We have compared our protein-level data to mRNA-level data from Kemmeren and colleagues (Kemmeren et al., Cell 2014), and we find very good agreement between the effects of gene perturbations on mRNA and protein levels when considering only genes with q < 0.05 and Log2FC > 0.5 in both studies (Pearson’s R = 0.79, p < 5.3e-15).
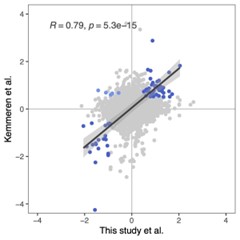
Gene perturbations with effects detected only on mRNA but not protein levels are enriched in genes with a role in “chromatin organization” (FDR = 0.01; as a background for the analysis, only the 1098 genes covered in both studies were considered). This suggests that perturbations of genes involved in chromatin organization tend to affect mRNA levels but are then buffered and do not lead to altered protein levels. There was no enrichment of functional annotations among gene perturbations with effects on protein levels but not mRNA levels.
We did not include these results in the manuscript because there are some limitations to the conclusions that can be drawn from these comparisons, including that our study has a relatively high number of false negatives, and that the genes perturbed in the Kemmeren et al. study were selected to play a role in gene regulation, meaning that differences in mRNA-vs-protein effects of perturbations are limited to this function, and other gene functions cannot be assessed.
- The finding that genes that affect only one or two proteins are enriched for roles in transcriptional regulation could be a consequence of 'only' looking at 10 proteins rather than a globally valid conclusion. Particularly as the 10 proteins were selected for diverse functions that are subject to distinct regulatory cascades. ('only' because I appreciate this was a lot of work.)
We agree with this, and we think it is clear in the abstract and the main text of the manuscript that here we studied 11 proteins. We made this point also more explicit in the discussion, so that it is clear for readers that the findings are based on the 11 proteins and may not extrapolate to the entire yeast proteome.
Reviewer #3 (Public Review):
This manuscript presents two main contributions. First, the authors modified a CRISPR base editing system for use in an important model organism: budding yeast. Second, they demonstrate the utility of this system by using it to conduct an extremely high throughput study the effects of mutation on protein abundance. This study confirms known protein regulatory relationships and detects several important new ones. It also reveals trends in the type of mutations that influence protein abundances. Overall, the findings are of high significance and the method appears to be extremely useful. I found the conclusions to be justified by the data.
One potential weakness is that some of the methods are not described in main body of the paper, so the reader has to really dive into the methods section to understand particular aspects of the study, for example, how the fitness competition was conducted.
We expanded the first section for better readability.
Another potential weakness is the comparison of this study (of protein abundances) to previous studies (of transcript abundances) was a little cursory, and left some open questions. For example, is it remarkable that the mutations affecting protein abundance are predominantly in genes involved in translation rather than transcription, or is this an expected result of a study focusing on protein levels?
We thank the reviewer for pointing out that this paragraph requires more explanation. We expanded it as follows: “Of these 29 genes, 21 (72%) have roles in protein translation—more specifically, in ribosome biogenesis and tRNA metabolism (FDR < 8.0e-4, Figure 5C). In contrast, perturbations that affect the abundance of only one or two of the eleven proteins mostly occur in genes with roles in transcription (e.g., GO:0006351, FDR < 1.3e-5). Protein biosynthesis entails both transcription and translation, and these results suggest that perturbations of translational machinery alter protein abundance broadly, while perturbations of transcriptional machinery can tune the abundance of individual proteins. Thus, genes with post-transcriptional functions are more likely to appear as hubs in protein regulatory networks, whereas genes with transcriptional functions are likely to show fewer connections.”
Overall, the strengths of this study far outweigh these weaknesses. This manuscript represents a very large amount of work and demonstrates important new insights into protein regulatory networks.
-
Evaluation Summary:
Schubert and coworkers report the development of a novel CRISPR-based screening method that allows probing interactions between (a large set of) specific mutations and the abundance of specific proteins, and, more generally, investigate the spectrum of effects that (point) mutations can have on protein abundance. This complements existing strategies for measuring effects of genetic perturbations on transcript levels, which is important as for some proteins mRNA and protein levels do not correlate well. The ability to measure proteins directly therefore promises to close an important gap in our understanding of the links between genotype and phenotype, and the strategy is broadly applicable beyond the current study.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the …
Evaluation Summary:
Schubert and coworkers report the development of a novel CRISPR-based screening method that allows probing interactions between (a large set of) specific mutations and the abundance of specific proteins, and, more generally, investigate the spectrum of effects that (point) mutations can have on protein abundance. This complements existing strategies for measuring effects of genetic perturbations on transcript levels, which is important as for some proteins mRNA and protein levels do not correlate well. The ability to measure proteins directly therefore promises to close an important gap in our understanding of the links between genotype and phenotype, and the strategy is broadly applicable beyond the current study.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 and Reviewer #2 agreed to share their name with the authors.)
-
Reviewer #1 (Public Review):
Overall, the science is sound and interesting, and the results are clearly presented. However, the paper falls in-between describing a novel method and studying biology. As a consequence, it is a bit difficult to grasp the general flow, central story and focus point. The study does uncover several interesting phenomena, but none are really studied in much detail and the novel biological insight is therefore a bit limited and lost in the abundance of observations. Several interesting novel interactions are uncovered, in particular for the SPS sensor and GAPDH paralogs, but these are not followed up on in much detail. The same can be said for the more general observations, eg the fact that different types of mutations (missense vs nonsense) in different types of genes (essential vs non-essential, housekeeping …
Reviewer #1 (Public Review):
Overall, the science is sound and interesting, and the results are clearly presented. However, the paper falls in-between describing a novel method and studying biology. As a consequence, it is a bit difficult to grasp the general flow, central story and focus point. The study does uncover several interesting phenomena, but none are really studied in much detail and the novel biological insight is therefore a bit limited and lost in the abundance of observations. Several interesting novel interactions are uncovered, in particular for the SPS sensor and GAPDH paralogs, but these are not followed up on in much detail. The same can be said for the more general observations, eg the fact that different types of mutations (missense vs nonsense) in different types of genes (essential vs non-essential, housekeeping vs. stress-regulated...) cause different effects.
This is not to say that the paper has no merit - far from it even. But, in its current form, it is a bit chaotic. Maybe there is simply too much in the paper? To me, it would already help if the authors would explicitly state that the paper is a "methods" paper that describes a novel technique for studying the effects of mutations on protein abundance, and then goes on to demonstrate the possibilities of the technology by giving a few examples of the phenomena that can be studied. The discussion section ends in this way, but it may be helpful if this was moved to the end of the introduction.
-
Reviewer #2 (Public Review):
Schubert et al. describe a new pooled screening strategy that combines protein abundance measurements of 11 proteins determined via FACS with genome-wide mutagenesis of stop codons and missense mutations (achieved via a base editor) in yeast. The method allows to identify genetic perturbations that affect steady state protein levels (vs transcript abundance), and in this way define regulators of protein abundance. The authors find that perturbation of essential genes more often alters protein abundance than of nonessential genes and proteins with core cellular functions more often decrease in abundance in response to genetic perturbations than stress proteins. Genes whose knockouts affected the level of several of the 11 proteins were enriched in protein biosynthetic processes while genes whose knockouts …
Reviewer #2 (Public Review):
Schubert et al. describe a new pooled screening strategy that combines protein abundance measurements of 11 proteins determined via FACS with genome-wide mutagenesis of stop codons and missense mutations (achieved via a base editor) in yeast. The method allows to identify genetic perturbations that affect steady state protein levels (vs transcript abundance), and in this way define regulators of protein abundance. The authors find that perturbation of essential genes more often alters protein abundance than of nonessential genes and proteins with core cellular functions more often decrease in abundance in response to genetic perturbations than stress proteins. Genes whose knockouts affected the level of several of the 11 proteins were enriched in protein biosynthetic processes while genes whose knockouts affected specific proteins were enriched for functions in transcriptional regulation. The authors also leverage the dataset to confirm known and identify new regulatory relationships, such as a link between the SDS amino acid sensor and the stress response gene Yhb1 or between Ras/PKA signalling and GAPDH isoenzymes Tdh1, 2, and 3. In addition, the paper contains a section on benchmarking of the base editor in yeast, where it has not been used before.
Strengths and weaknesses of the paper:
The authors establish the BE3 base editor as a screening tool in S. cerevisiae and very thoroughly benchmark its functionality for single edits and in different screening formats (fitness and FACS screening). This will be very beneficial for the yeast community.The strategy established here allows measuring the effect of genetic perturbations on protein abundances in highly complex libraries. This complements capabilities for measuring effects of genetic perturbations on transcript levels, which is important as for some proteins mRNA and protein levels do not correlate well. The ability to measure proteins directly therefore promises to close an important gap in determining all their regulatory inputs. The strategy is furthermore broadly applicable beyond the current study. All experimental procedures are very well described and plasmids and scripts are openly shared, maximizing utility for the community.
There is a good balance between global analyses aimed at characterizing properties of the regulatory network and more detailed analyses of interesting new regulatory relationships. Some of the key conclusions are further supported by additional experimental evidence, which includes re-making specific mutations and confirming their effects on protein levels by mass spectrometry.
The conclusions of the paper are mostly well supported, but I am missing some analyses on reproducibility and potential confounders and some of the data analysis steps should be clarified.
The paper starts on the premise that measuring protein levels will identify regulators and regulatory principles that would not be found by measuring transcripts, but since the findings are not discussed in light of studies looking at mRNA levels it is unclear how the current study extends knowledge regarding the regulatory inputs of each protein.
Specific comments regarding data analysis, reproducibility, confounders:
The authors use the number of unique barcodes per guide RNA rather than barcode counts to determine fold-changes. For reliable fold changes the number of unique barcodes per gRNA should then ideally be in the 100s for each guide, is that the case? It would also be important to show the distribution of the number of barcodes per gRNA and their abundances determined from read counts. I could imagine that if the distribution of barcodes per gRNA or the abundance of these barcodes is highly skewed (particularly if there are many barcodes with only few reads) that could lead to spurious differences in unique barcode number between the high and low fluorescence pool. I imagine some skew is present as is normal in pooled library experiments. The fold-changes in the control pools could show whether spurious differences are a problem, but it is not clear to me if and how these controls are used in the protein screen.I like the idea of using an additional barcode (plasmid barcode) to distinguish between different cells with the same gRNA - this would directly allow to assess variability and serve as a sort of replicate within replicate. However, this information is not leveraged in the analysis. It would be nice to see an analysis of how well the different plasmid barcodes tagging the same gRNA agree (for fitness and protein abundance), to show how reproducible and reliable the findings are.
From Fig 1 and previous research on base editors it is clear that mutation outcomes are often heterogeneous for the same gRNA and comprise a substantial fraction of wild-type alleles, alleles where only part of the Cs in the target window or where Cs outside the target window are edited, and non C-to-T edits. How does this reflect on the variability of phenotypic measurements, given that any barcode represents a genetically heterogeneous population of cells rather than a specific genotype? This would be important information for anyone planning to use the base editor in future.
How common are additional mutations in the genome of these cells and could they confound the measured effects? I can think of several sources of additional mutations, such as off-target editing, edits outside the target window, or when 2 gRNA plasmids are present in the same cell (both target windows obtain edits). Could some of these events explain the discrepancy in phenotype for two gRNAs that should make the same mutation (Fig S4)? Even though BE3 has been described in mammalian cells, an off-target analysis would be desirable as there can be substantial differences in off-target behavior between cell types and organisms.
In the protein screen normalization uses the total unique barcode counts. Does this efficiently correct for differences from sequencing (rather than total read counts or other methods)? It would be nice to see some replicate plots for the analysis of the fitness as well as the protein screen to be able to judge that.
In the main text the authors mention very high agreement between gRNAs introducing the same mutation but this is only based on 20 or so gRNA pairs; for many more pairs that introduce the same mutation only one reaches significance, and the correlation in their effects is lower (Fig S4). It would be better to reflect this in the text directly rather than exclusively in the supplementary information.
When the different gRNAs for a targeted gene are combined, instead of using an averaged measure of their effects the authors use the largest fold-change. This seems not ideal to me as it is sensitive to outliers (experimental error or background mutations present in that strain).
Phenotyping is performed directly after editing, when the base editor is still present in the cells and could still interact with target sites. I could imagine this could lead to reduced levels of the proteins targeted for mutagenesis as it could act like a CRISPRi transcriptional roadblock. Could this enhance some of the effects or alter them in case of some missense mutations?
I feel that the main text does not reflect the actual editing efficiency very well (the main numbers I noticed were 95% C to T conversion and 89% of these occurring in a specific window). More informative for interpreting the results would be to know what fraction of the alleles show an edit (vs wild-type) and how many show the 'complete' edit (as the authors assume 100% of the genotypes generated by a gRNA to be conversion of all Cs to Ts in the target window). It would be important to state in the main text how variable this is for different gRNAs and what the typical purity of editing outcomes is.
Comments regarding findings:
It would be nice to see a comparison of the results to the effects of ~1500 yeast gene knockouts on cellular transcriptomes (https://doi.org/10.1016/j.cell.2014.02.054). This would show where the current study extends established knowledge regarding the regulatory inputs of each protein and highlight the importance of directly measuring protein levels. This would be particularly interesting for proteins whose abundance cannot be predicted well from mRNA abundance.The finding that genes that affect only one or two proteins are enriched for roles in transcriptional regulation could be a consequence of 'only' looking at 10 proteins rather than a globally valid conclusion. Particularly as the 10 proteins were selected for diverse functions that are subject to distinct regulatory cascades. ('only' because I appreciate this was a lot of work.)
-
Reviewer #3 (Public Review):
This manuscript presents two main contributions. First, the authors modified a CRISPR base editing system for use in an important model organism: budding yeast. Second, they demonstrate the utility of this system by using it to conduct an extremely high throughput study the effects of mutation on protein abundance. This study confirms known protein regulatory relationships and detects several important new ones. It also reveals trends in the type of mutations that influence protein abundances. Overall, the findings are of high significance and the method appears to be extremely useful. I found the conclusions to be justified by the data.
One potential weakness is that some of the methods are not described in main body of the paper, so the reader has to really dive into the methods section to understand …
Reviewer #3 (Public Review):
This manuscript presents two main contributions. First, the authors modified a CRISPR base editing system for use in an important model organism: budding yeast. Second, they demonstrate the utility of this system by using it to conduct an extremely high throughput study the effects of mutation on protein abundance. This study confirms known protein regulatory relationships and detects several important new ones. It also reveals trends in the type of mutations that influence protein abundances. Overall, the findings are of high significance and the method appears to be extremely useful. I found the conclusions to be justified by the data.
One potential weakness is that some of the methods are not described in main body of the paper, so the reader has to really dive into the methods section to understand particular aspects of the study, for example, how the fitness competition was conducted. Another potential weakness is the comparison of this study (of protein abundances) to previous studies (of transcript abundances) was a little cursory, and left some open questions. For example, is it remarkable that the mutations affecting protein abundance are predominantly in genes involved in translation rather than transcription, or is this an expected result of a study focusing on protein levels?
Overall, the strengths of this study far outweigh these weaknesses. This manuscript represents a very large amount of work and demonstrates important new insights into protein regulatory networks.
-
