The NAD+ precursor NMN activates dSarm to trigger axon degeneration in Drosophila
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
Regulation of NAD and its intermediary metabolites is of critical importance in axon degeneration and in neurodegenerative disease. In mammals, the SARM1 NADase has been shown to be a metabolic sensor activated by an increase in the NMN/NAD+ ratio and SARM1 activation then leads to catastrophic energetic collapse and axon degeneration in disease and injury. This manuscript clarifies the role of NMN in activating the axon degeneration trigger dSARM in Drosophila. The authors analyze the signaling role of NMN, a NAD precursor metabolite involved in injury-induced axon degeneration, by overexpressing NMN-D, a prokaryotic enzyme that consumes NMN, using a stabilized version allowing for prolonged NMN depletion, and find that it is strongly protective in several in vivo injury paradigms in flies. This paper will be of interest to those in the neurodegeneration/axon injury field in general as an extensive set of optimized reagents is presented, confirming the crucial role of for exploring NAD-related axon degenerative pathways, and providing tools for neuroscientists to use Drosophila as a model for neurodegenerative research.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #2 agreed to share their name with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Axon degeneration contributes to the disruption of neuronal circuit function in diseased and injured nervous systems. Severed axons degenerate following the activation of an evolutionarily conserved signaling pathway, which culminates in the activation of SARM1 in mammals to execute the pathological depletion of the metabolite NAD + . SARM1 NADase activity is activated by the NAD + precursor nicotinamide mononucleotide (NMN). In mammals, keeping NMN levels low potently preserves axons after injury. However, it remains unclear whether NMN is also a key mediator of axon degeneration and dSarm activation in flies. Here, we demonstrate that lowering NMN levels in Drosophila through the expression of a newly generated prokaryotic NMN-Deamidase (NMN-D) preserves severed axons for months and keeps them circuit-integrated for weeks. NMN-D alters the NAD + metabolic flux by lowering NMN, while NAD + remains unchanged in vivo. Increased NMN synthesis by the expression of mouse nicotinamide phosphoribosyltransferase (mNAMPT) leads to faster axon degeneration after injury. We also show that NMN-induced activation of dSarm mediates axon degeneration in vivo. Finally, NMN-D delays neurodegeneration caused by loss of the sole NMN-consuming and NAD + -synthesizing enzyme dNmnat. Our results reveal a critical role for NMN in neurodegeneration in the fly, which extends beyond axonal injury. The potent neuroprotection by reducing NMN levels is similar to the interference with other essential mediators of axon degeneration in Drosophila .
Article activity feed
-

-

Author Response
Reviewer #2 (Public Review):
Regulation of NAD and its intermediary metabolites is of critical importance in axon degeneration and neurodegenerative disease. Mounting evidence supports a scenario in which low NAD, and high NMN triggers axon degeneration by competitive allosteric inhibition/activation of SARM1. Strategies to increase NAD levels and/or lower NMN levels provide neuroprotection in a variety of contexts. NAD metabolism is a partially conserved process, however, there are key differences in pathway routes and dynamics between model organisms used for NAD research (yeast, worm, fly, zebrafish, mouse/mammalian systems). Drosophila is a key model organism for axon degenerative research based on its ease of use and range of available genetic tools, in addition, the effector of axon degeneration - SARM1 - was …
Author Response
Reviewer #2 (Public Review):
Regulation of NAD and its intermediary metabolites is of critical importance in axon degeneration and neurodegenerative disease. Mounting evidence supports a scenario in which low NAD, and high NMN triggers axon degeneration by competitive allosteric inhibition/activation of SARM1. Strategies to increase NAD levels and/or lower NMN levels provide neuroprotection in a variety of contexts. NAD metabolism is a partially conserved process, however, there are key differences in pathway routes and dynamics between model organisms used for NAD research (yeast, worm, fly, zebrafish, mouse/mammalian systems). Drosophila is a key model organism for axon degenerative research based on its ease of use and range of available genetic tools, in addition, the effector of axon degeneration - SARM1 - was first identified in the fly. As Drosophila has some key differences in the NAD synthesis pathways to mammalian systems it is important to test and develop tools to enable exploration of these pathways on the fly. Llobet Rosell and colleagues have developed clear and demonstrable tools in Drosophila for exploring NAD-related axon degenerative pathways by modulating the use of NMN via the addition of NMN consuming and NMN generating enzymes. They utilize Drosophila genetics to adequately support the claims made in the manuscript. Importantly, the authors well-demonstrate that consuming NMN through an alternate route to NaMN provides neuroprotection and that the neuroprotective components of low NMN are upstream of SARM1. These should be useful tools for neuroscientists in the future to use Drosophila for neurodegenerative research.
Strengths:
• Clear demonstration that low NMN provides neuroprotection using novel, stable, enzymatic depletion of NMN (to NaMN).
• Development of a novel Drosophila tool (NMN-D transgenics) to explore NMN metabolism in vivo, including a stabilized version to permit chronic NMN depletion.
• Metabolomic profiles across the pathway to show all pathway changes (not just isolated NMN or NAD assays). • Neurodegenerative assays that include both histological outcomes (axon degeneration) but also circuitry/functional outcomes. Data from both series of experiments all support each other.
• Assessment of other known potent axon degenerative genes via genetics in combination with the tools developed. • Staging of the molecular processes by strategic ablation of the inhibitory ARM domain on SARM1 (dSarm deltaARM). These experiments suggest that low NAD AND high NMN (i.e. ratio between the two) is the critical factor that drives axon degeneration. Once NAD is low, axon degeneration cannot be recovered by further lowering of NMN. The dSarm delta-ARM and dnmnat sgRNAs experiments support a hypothesis in that (high) NMN triggers, but doesn't, execute axon degeneration.
We appreciate his recognition of the quality of our research.
Weaknesses:
• The authors use murine NAMPT (mNAMPT) to increase NMN. The degeneration assays support the hypotheses made, yet mNAMPT doesn't actually increase NMN. Thus it is unclear in this setting whether mNAMPT promotes axon degeneration by an NMN-related mechanism or through another route. It is also unclear as to why the murine form was chosen versus a human or other orthologues, or changing the metabolism of the intrinsic pathway (NR and NRK).
Why mNAMPT:
We decided to use mouse NAMPT (mNAMPT) because it was readily available by Giuseppe Orsomando (Amici et al., 2017), and because we did not have access to human NAMPT (hNAMPT).
We agree with the observation that under physiological conditions, the expression of mNAMPT does not change NMN. However, we argue that after injury, once dNmnat is degraded, the additional NMN synthesis provided by mNAMPT expression (in addition to dNrk), leads to a faster NMN accumulation. It is supported by the observation that NMNAT2 is more labile than NAMPT in mammals (Gilley and Coleman, 2010; Stefano et al., 2015).• The authors use metabolic profiling to look at the individual metabolites during axon degenerative evens and treatments however it is unclear if any of these proteins or genes change as a consequence. This is likely not important for understanding the findings however, might be helpful in explaining the mNAMPT data.
We agree with the idea to test whether there is a change induced at the mRNA or protein level when the metabolic flux is altered. To do this, first, we measured the relative expression levels of axon death and NAD+ synthesis genes (Figure 2 – figure supplement 1B). Then, we measured potential changes upon mNAMPT expression (Figure 4 – figure supplement 1). Importantly, while the Gal4-driven expression resulted in an increase of relative mNAMPT transcript abundance from 30 to 12’000, the change observed in the other genes was not notable. Importantly, compared to Actin–Gal4, dnrk is 2-fold lower in UAS-mNAMPT and Actin > mNAMPT backgrounds (control vs. experiment, respectively). Thus, overall, there appears to be no change in mRNAs of either axon death or NAD+ synthesis genes.
In the results, we changed the text accordingly:
"We then tested the effect of mNAMPT on the NAD+ metabolic flux in vivo. Surprisingly, NAM, NMN, and NAD+ levels remained unchanged under physiological conditions (Figure 4C). However, we noticed 3-fold higher NR and a moderate but significant elevation of ADPR and cADPR levels upon mNAMPT overexpression (Figure 4C). We also asked whether mNAMPT impacts on NAD+ homeostasis thereby altering the expression of axon death or NAD+ synthesis genes. Besides the expected significant increase in the Gal4-mediated expression of mNAMPT, we did not observe any notable changes at the mRNA level (Figure 4 – figure supplement 1)."
• The authors repeatedly introduce a novel PncC antibody. However, no details on this, its generation, or its testing are found within the manuscript as presented. The antibody detects with several bands. The authors speculate that this could be a degradation product but nothing substantial is shown.
In Materials and methods, we added a new section:
"PncC antibody generation Rabbit anti-PncC antibodies were generated by Lubioscience under a proprietary protocol. The immunogen used was purified from Escherichia coli, strain K12, corresponding to the full protein sequence of NMN-D. The amino acid sequence is the following: MTDSELMQLSEQVGQALKARGATVTTAESCTGGWVAKVITDIAGSSAWFERGFVTYSNEAKAQMIGVREETLAQHGAVSEPVVVEMAIGALKAARADYAVSISGIAGPDGGSEEKPVGVWFAFATARGEGITRRECFSGDRDAVRRQAT AYALQTLWQQFLQNT"
We also updated the results referencing it.
"We found that both wild-type and enzymatically dead NMN-D enzymes are equally expressed in S2 cells, as detected by newly generated PncC antibodies (Materials & Methods, Figure 1–figure supplement 2). Notably, we observed two immunoreactivities per lane, with the lower band being a potential degradation product."
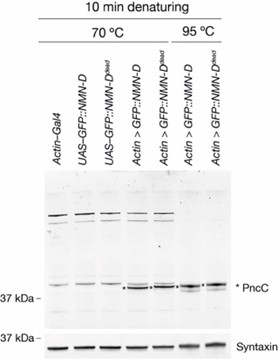
In addition, we now provide evidence why we believe that the upper band is NMN-D, while the lower one is a degradation product. In the figure attached below, the samples of the first five lanes were denatured at 70 °C, while the samples of the last two lanes were denatured at 95 °C (each for 10 min, respectively). The resulting Western blot shows that at 70 °C, there is more unspecific background, but no lower degradation product, while at 95 °C, the background is drastically reduced; however, there is a lower degradation product appearing. NMN-D is indicated by an asterisk. We feel that it is important to show this data here in the rebuttal. But we feel that it would add confusion to the readers in the manuscript.
• Olfactory receptor neuron degeneration assays are shown in Fig1 but no data is presented with it to support the images.
We agree that a quantification would support our observation. However, it is difficult to precisely quantify individual axons in the ORN injury assay, for two main reasons:
Severed axons are often bundled, thus the exact number cannot be scored.
Due to the removal of the cell body, the axonal GFP intensity decreases over time, due to the absence of mCD8::GFP synthesis. It adds another level of difficulty. Nevertheless, we added numbers to each example in Figure 1E and D, where we quantified the % of brains where severed preserved axons were observed, similar to Figure 2 in (MacDonald et al., 2006).
In the results section, we changed the text as indicated below:
"We extended the ORN injury assay and found preservation at 10, 30, and 50 dpa (Figure 1E). While quantifying the precise number of axons is technically not feasible, severed preserved axons were observed in all 10, 30, and 50 dpa brains, albeit fewer at later time points (MacDonald et al., 2006). Thus, high levels of NMN-D confer robust protection of severed axons for multiple neuron types for the entire lifespan of Drosophila."
In the Figure 1 legend, we changed the text accordingly:
"D Low NMN results in severed axons of olfactory receptor neurons that remain morphologically preserved at 7 dpa. Examples of control and 7 dpa (arrows, site of unilateral ablation). Lower right, % of brains with severed preserved axon fibers. E Low NMN results in severed axons that remain morphologically preserved for 50 days. Representative pictures of 10, 30, and 50 dpa, from a total of 10 brains imaged for each condition (arrows, site of unilateral ablation). Lower right, % of brains with severed preserved axon fibers."
-
Evaluation Summary:
Regulation of NAD and its intermediary metabolites is of critical importance in axon degeneration and in neurodegenerative disease. In mammals, the SARM1 NADase has been shown to be a metabolic sensor activated by an increase in the NMN/NAD+ ratio and SARM1 activation then leads to catastrophic energetic collapse and axon degeneration in disease and injury. This manuscript clarifies the role of NMN in activating the axon degeneration trigger dSARM in Drosophila. The authors analyze the signaling role of NMN, a NAD precursor metabolite involved in injury-induced axon degeneration, by overexpressing NMN-D, a prokaryotic enzyme that consumes NMN, using a stabilized version allowing for prolonged NMN depletion, and find that it is strongly protective in several in vivo injury paradigms in flies. This paper will be of …
Evaluation Summary:
Regulation of NAD and its intermediary metabolites is of critical importance in axon degeneration and in neurodegenerative disease. In mammals, the SARM1 NADase has been shown to be a metabolic sensor activated by an increase in the NMN/NAD+ ratio and SARM1 activation then leads to catastrophic energetic collapse and axon degeneration in disease and injury. This manuscript clarifies the role of NMN in activating the axon degeneration trigger dSARM in Drosophila. The authors analyze the signaling role of NMN, a NAD precursor metabolite involved in injury-induced axon degeneration, by overexpressing NMN-D, a prokaryotic enzyme that consumes NMN, using a stabilized version allowing for prolonged NMN depletion, and find that it is strongly protective in several in vivo injury paradigms in flies. This paper will be of interest to those in the neurodegeneration/axon injury field in general as an extensive set of optimized reagents is presented, confirming the crucial role of for exploring NAD-related axon degenerative pathways, and providing tools for neuroscientists to use Drosophila as a model for neurodegenerative research.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #2 agreed to share their name with the authors.)
-
Reviewer #1 (Public Review):
In this study, the authors overexpress GFP-tagged NMN-D, a prokaryotic enzyme that consumes NMN, and find that it is strongly protective in several in vivo injury paradigms in flies. This is an important finding that clarifies previously published work, which found that an untagged NMN-D construct provided only weak axon protection (Hsu et al., 2021). The authors of the current manuscript argue convincingly that the previous result stemmed from the low stability of the untagged variant. Llobet Rossell et al. also use a very nice grooming assay for synaptic connectivity following axotomy to demonstrate that NMN-D overexpression maintains synaptic connectivity. Further pointing to NMN as a crucial regulator of dSARM activation, they show that increasing NMN levels by increasing NMN synthesis through mNAMPT …
Reviewer #1 (Public Review):
In this study, the authors overexpress GFP-tagged NMN-D, a prokaryotic enzyme that consumes NMN, and find that it is strongly protective in several in vivo injury paradigms in flies. This is an important finding that clarifies previously published work, which found that an untagged NMN-D construct provided only weak axon protection (Hsu et al., 2021). The authors of the current manuscript argue convincingly that the previous result stemmed from the low stability of the untagged variant. Llobet Rossell et al. also use a very nice grooming assay for synaptic connectivity following axotomy to demonstrate that NMN-D overexpression maintains synaptic connectivity. Further pointing to NMN as a crucial regulator of dSARM activation, they show that increasing NMN levels by increasing NMN synthesis through mNAMPT overexpression accelerates injury-induced axon degeneration. They provide the support that NMN-D and mNAMPT overexpression are having the expected effect on NAD+ metabolic flux via LC-MS/MS. Finally, they provide evidence that a dSARM variant that cannot bind NMN does not rescue the dSARM LOF phenotype indicating that NMN exerts its effects through dSARM.
This study is carefully done and the evidence that NMN levels are a critical determinant of axon degeneration is strong. The combination of in vivo degeneration assays, metabolomics, and behavioral assays provides confidence in the results. For the most part, the conclusions of the study are well-supported by the results. The claim that the protection afforded by low NMN levels is as strong as the loss of SARM is justified. The authors repeatedly state that protection provided by low NMN is "even stronger" than that of essential mediators of axon degeneration, which doesn't really make sense given that all their data support the hypothesis that low NMN protects because it blocks dSARM activation. However, the primary weakness of this study is that the novelty of this work comes almost entirely from showing that the NMN/NAD+ ratio is determinative for axon degeneration in Drosophila; it has already been shown both in mammalian cultured neurons and in vivo.
-
Reviewer #2 (Public Review):
Regulation of NAD and its intermediary metabolites is of critical importance in axon degeneration and neurodegenerative disease. Mounting evidence supports a scenario in which low NAD, and high NMN triggers axon degeneration by competitive allosteric inhibition/activation of SARM1. Strategies to increase NAD levels and/or lower NMN levels provide neuroprotection in a variety of contexts. NAD metabolism is a partially conserved process, however, there are key differences in pathway routes and dynamics between model organisms used for NAD research (yeast, worm, fly, zebrafish, mouse/mammalian systems). Drosophila is a key model organism for axon degenerative research based on its ease of use and range of available genetic tools, in addition, the effector of axon degeneration - SARM1 - was first identified in …
Reviewer #2 (Public Review):
Regulation of NAD and its intermediary metabolites is of critical importance in axon degeneration and neurodegenerative disease. Mounting evidence supports a scenario in which low NAD, and high NMN triggers axon degeneration by competitive allosteric inhibition/activation of SARM1. Strategies to increase NAD levels and/or lower NMN levels provide neuroprotection in a variety of contexts. NAD metabolism is a partially conserved process, however, there are key differences in pathway routes and dynamics between model organisms used for NAD research (yeast, worm, fly, zebrafish, mouse/mammalian systems). Drosophila is a key model organism for axon degenerative research based on its ease of use and range of available genetic tools, in addition, the effector of axon degeneration - SARM1 - was first identified in the fly. As Drosophila has some key differences in the NAD synthesis pathways to mammalian systems it is important to test and develop tools to enable exploration of these pathways on the fly. Llobet Rosell and colleagues have developed clear and demonstrable tools in Drosophila for exploring NAD-related axon degenerative pathways by modulating the use of NMN via the addition of NMN consuming and NMN generating enzymes. They utilize Drosophila genetics to adequately support the claims made in the manuscript. Importantly, the authors well-demonstrate that consuming NMN through an alternate route to NaMN provides neuroprotection and that the neuroprotective components of low NMN are upstream of SARM1. These should be useful tools for neuroscientists in the future to use Drosophila for neurodegenerative research.
Strengths:
• Clear demonstration that low NMN provides neuroprotection using novel, stable, enzymatic depletion of NMN (to NaMN).
• Development of a novel Drosophila tool (NMN-D transgenics) to explore NMN metabolism in vivo, including a stabilized version to permit chronic NMN depletion.
• Metabolomic profiles across the pathway to show all pathway changes (not just isolated NMN or NAD assays).
• Neurodegenerative assays that include both histological outcomes (axon degeneration) but also circuitry/functional outcomes. Data from both series of experiments all support each other.
• Assessment of other known potent axon degenerative genes via genetics in combination with the tools developed.
• Staging of the molecular processes by strategic ablation of the inhibitory ARM domain on SARM1 (dSarm delta-ARM). These experiments suggest that low NAD AND high NMN (i.e. ratio between the two) is the critical factor that drives axon degeneration. Once NAD is low, axon degeneration cannot be recovered by further lowering of NMN. The dSarm delta-ARM and dnmnat sgRNAs experiments support a hypothesis in that (high) NMN triggers, but doesn't, execute axon degeneration.Weaknesses:
• The authors use murine NAMPT (mNAMPT) to increase NMN. The degeneration assays support the hypotheses made, yet mNAMPT doesn't actually increase NMN. Thus it is unclear in this setting whether mNAMPT promotes axon degeneration by an NMN-related mechanism or through another route. It is also unclear as to why the murine form was chosen versus a human or other orthologues, or changing the metabolism of the intrinsic pathway (NR and NRK).
• The authors use metabolic profiling to look at the individual metabolites during axon degenerative evens and treatments however it is unclear if any of these proteins or genes change as a consequence. This is likely not important for understanding the findings however, might be helpful in explaining the mNAMPT data.
• The authors repeatedly introduce a novel PncC antibody. However, no details on this, its generation, or its testing are found within the manuscript as presented. The antibody detects with several bands. The authors speculate that this could be a degradation product but nothing substantial is shown.
• Olfactory receptor neuron degeneration assays are shown in Fig1 but no data is presented with it to support the images. -
Reviewer #3 (Public Review):
Llobet-Rosell et al. use Drosophila to decipher the relationships between factors in the Wallerian degeneration pathway and the metabolite NMN, an activator of the central pathway enzyme dSarm. NMN had previously been proposed to be a crucial regulator of Sarm, but there was a shortage of good in vivo evidence, especially in the crucial Drosophila system. The authors addressed this here by generating optimized fly lines, including a strongly expressing transgenic line for the NMN-consuming enzyme NMN-deamidase (NMNd). This variant conferred extremely strong protection against degeneration both in morphological and functional studies, thus confirming the key role of NMN as an activator of the degeneration pathway. They also confirm that NMNd alters NMN/NAD metabolism using mass spec of Drosophila heads, and …
Reviewer #3 (Public Review):
Llobet-Rosell et al. use Drosophila to decipher the relationships between factors in the Wallerian degeneration pathway and the metabolite NMN, an activator of the central pathway enzyme dSarm. NMN had previously been proposed to be a crucial regulator of Sarm, but there was a shortage of good in vivo evidence, especially in the crucial Drosophila system. The authors addressed this here by generating optimized fly lines, including a strongly expressing transgenic line for the NMN-consuming enzyme NMN-deamidase (NMNd). This variant conferred extremely strong protection against degeneration both in morphological and functional studies, thus confirming the key role of NMN as an activator of the degeneration pathway. They also confirm that NMNd alters NMN/NAD metabolism using mass spec of Drosophila heads, and then use Drosophila genetics to show that dSarm is the crucial NMN target. In a reverse experiment, the authors also use overexpression of murine NAMPT, an NMN-producing enzyme, to speed up degeneration. As in mammals, NMNd delays degeneration induced by loss of Nmnat.
A clear strength of the fly system is the degree of rescue conferred by the optimized NMN-D reagent which essentially establishes NMN as a crucial regulator in the pathway. The rigor of experimentation is also very high. Essentially all reagents are optimized, and most conclusions are backed by complementary analyses. The manuscript also nicely describes a metabolomic analysis of NAD biosynthetic pathways from fly heads.
-
