Identification of phenotypically, functionally, and anatomically distinct stromal niche populations in human bone marrow based on single-cell RNA sequencing
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
The manuscript by Li and coworkers characterizes sorted human non-hematopoietic bone marrow cells by scRNA-seq and predicts their lineage relationships and possible interactions with mature and immature hematopoietic cells. Transcriptionally-different stromal cell subsets are identified, and their lineage relationships, cell-cell interactions and possible specialized functions are inferred or predicted from in-silico studies, paving the way for future functional and validation studies. This resource significantly adds to the current understanding human non-hematopoietic bone marrow stromal cells and their hematopoietic regulatory functions.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 and Reviewer #2 agreed to share their name with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Hematopoiesis is regulated by the bone marrow (BM) stroma. However, cellular identities and functions of the different BM stromal elements in humans remain poorly defined. Based on single-cell RNA sequencing (scRNAseq), we systematically characterized the human non-hematopoietic BM stromal compartment and we investigated stromal cell regulation principles based on the RNA velocity analysis using scVelo and studied the interactions between the human BM stromal cells and hematopoietic cells based on ligand-receptor (LR) expression using CellPhoneDB. scRNAseq led to the identification of six transcriptionally and functionally distinct stromal cell populations. Stromal cell differentiation hierarchy was recapitulated based on RNA velocity analysis and in vitro proliferation capacities and differentiation potentials. Potential key factors that might govern the transition from stem and progenitor cells to fate-committed cells were identified. In situ localization analysis demonstrated that different stromal cells were localized in different niches in the bone marrow. In silico cell-cell communication analysis further predicted that different stromal cell types might regulate hematopoiesis through distinct mechanisms. These findings provide the basis for a comprehensive understanding of the cellular complexity of the human BM microenvironment and the intricate stroma-hematopoiesis crosstalk mechanisms, thus refining our current view on human hematopoietic niche organization.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
This is an awesome comprehensive manuscript. Authors start by sorting putative stromal cellcontaining BM non-hematopoietic (CD235a-/CD45-) plus additional CD271+/CD235a/CD45- populations to identify nine individual stromal identities by scRNA-seq. The dual sorting strategy is a clever trick as it enriches for rare stromal (progenitor) cell signals but may suffer a certain bias towards CD271+ stromal progenitors. The lack of readable signatures already among CD45-/CD45- sorts might argue against this fear. This reviewer would appreciate a brief discussion on number & phenotype of putative additional MSSC phenotypes in light of the fact that the majority of 'blood lineage(s)'-negative scRNA-seq signatures identified blood cell progenitor identities (glycophorin A-negative & leukocyte common …
Author Response
Reviewer #1 (Public Review):
This is an awesome comprehensive manuscript. Authors start by sorting putative stromal cellcontaining BM non-hematopoietic (CD235a-/CD45-) plus additional CD271+/CD235a/CD45- populations to identify nine individual stromal identities by scRNA-seq. The dual sorting strategy is a clever trick as it enriches for rare stromal (progenitor) cell signals but may suffer a certain bias towards CD271+ stromal progenitors. The lack of readable signatures already among CD45-/CD45- sorts might argue against this fear. This reviewer would appreciate a brief discussion on number & phenotype of putative additional MSSC phenotypes in light of the fact that the majority of 'blood lineage(s)'-negative scRNA-seq signatures identified blood cell progenitor identities (glycophorin A-negative & leukocyte common antigen-negative). The nine stromal cell entities share the CXCL12, VCAN, LEPR main signature. Perhaps the authors could speculate if future studies using VCAN or LEPRbased sort strategies could identify additional stromal progenitor identities?
We would like to thank the reviewer for critically evaluating our work and for the generally positive evaluation of the paper. We apologize for delayed resubmission as it took a long time for a specific antibody to arrive to complete the confocal microscopy analyses.
The reviewer asks for a brief discussion on the cell numbers and phenotypes of MSSC phenotypes. The cell numbers and percentages of MSSC in sorted CD45low/-CD235a- and CD45low/-CD235a-CD271+ cells can be found in Supplementary File 3 and we have added a summary of the phenotypes of MSSC in the new Supplementary File 7.
Due to the extremely low frequency of stromal cells in human bone marrow, we chose a sorting strategy that also included CD45low cells (Fig 1A) to ensure that no stromal cells were excluded from the analysis. Although stromal elements are certainly enriched using this approach, the CD45low population contains several different hematopoietic cell types. These include CD34+ HSPCs which are characterized by low CD45 expression2, as well as the CD45low-expressing fractions of other hematopoietic cell populations such as B cells, T cells, NK cells, megakaryocytes, monocytes, dendritic cells, and granulocytes. Furthermore, CD235a- late-stage erythroid progenitors, which are negative for CD45, are represented as well. Of note, our data are consistent with previously reported murine studies showing the presence of a number of hematopoietic populations in CD45- cells, which accounted for the majority of CD45-Ter119-CD31- murine BM cells3,4. However, despite a certain enrichment of stromal elements in the CD45low cell fraction, frequencies were still too low to allow for a detailed analysis of this important bone marrow compartment. This prompted us to adopt the stromal cell-enrichment strategy as described in the manuscript to achieve a better resolution of the stromal compartment. In fact, sorting based on CD45low/-CD235a-CD271+ allowed us to sufficiently enrich bone marrow stromal cells to be clearly detectable in scRNAseq analysis. According to the reviewer’s suggestion, a brief discussion on this issue is now included in the Discussion (page 28, lines 10-15).
The reviewer also suggested using VCAN or LEPR-based sorting strategy to identify additional stromal identities in future studies.
However, as an extracellular matrix protein, FACS analysis of cellular VCAN expression can only be achieved based on its intracellular expression after fixation and permeabilization5,6. Additionally, while VCAN is highly and ubiquitously expressed by stromal clusters, VCAN is also expressed by monocytes (cluster 36). Therefore, VCAN is not an optimal marker to isolate viable stromal cells.
LEPR is the marker that was reported to identify the majority of colony-forming cells in adult murine bone marrow7. We have previously reported that the majority of human adult bone marrow CFU-Fs is contained in the LEPR+ fraction 8. In our current scRNAseq surface marker profiling analysis, group A cells showed high expression of several canonical stromal markers including VCAM1, PDGFRB, ENG (CD73), as well as LEPR (Fig. 4A). However, the four stromal clusters in Group A could not be separated based on the expression of LEPR. Therefore, we chose not to use LEPR as a marker to prospectively isolate the different stromal cell types.
The authors furthermore localized CD271+, CD81+ and NCAM/CD56+ cells in BM sections in situ. Finally, referring to the strong background of the group in HSC research, in silico prediction by CellPhoneDB identified a wide range of interactions between stromal cells and hematopoietic cells. Evidence for functional interdependence of FCU-F forming cells is completing the novel and more clear bone marrow stromal cell picture.
We thank the reviewer for the positive comments.
An illustrative abstract naming the top9 stromal identities in their top4 clusters by their "top10 markers" + functions would be highly appreciated.
We thank the reviewer for the suggestion. A summary of the characteristics of stromal clusters is now shown in the new Supplementary File 7, which we hope matches the reviewer’s expectations.
Reviewer #2 (Public Review):
Knowledge about composition and function of the different subpopulations of the hematopoietic niche of the BM is limited. Although such knowledge about the mouse BM has been accumulating in recent years, a thorough study of the human BM still needs to be performed. The present manuscript of Li and coworkers fills this gap by performing single cell RNA sequencing (scRNAseq) on control BM as well as CD271+ BM cells enriched for non-hematopoietic niche cells.
We apologize for delayed resubmission as it took a long time for a specific antibody to arrive to complete the confocal microscopy analyses. We thank the reviewer for the critical expert review and overall positive comments.
Based on their scRNAseq, the authors propose 41 different BM cell populations, ten of which represented non-hematopoietic cells, including one endothelial cell cluster. The nine remaining skeletal subpopulations were subdivided into multipotent stromal stem cells (MSSC), four distinct populations of osteoprogenitors, one cluster of osteoblasts and three clusters of pre-fibroblasts. Using bioinformatic tools, the authors then compare their results and divisions of subpopulations to some previously published work from others and attempt to delineate lineage relationships using RNA velocity analyses. From these, they propose different paths from which MSSC enter the progenitor stages, and might differentiate into pre-osteoblasts and -fibroblasts.
It is of interest to note, that apparently adipo-primed cells may also differentiate into osteolineage cells, something that should be further explored or validated. Furthermore, although this analysis yields a large adipo-primed populations, pre-adipocytes and mature adipocytes appear not to be included in the data set the authors used, which should also be explained.
We thank the reviewer for this comment. We chose to annotate Cluster 5 as adipoprimed cluster based on the higher expression of adipogenic differentiation markers as well as a group of stress-related transcription factors (FOS, FOSB, JUNB, EGR1) (Fig. 2B-C, Figure 2-figure supplement 1C) some of which had been shown to mark bone marrow adipogenic progenitors1. Although at considerably lower levels compared to adipogenic genes, osteogenic genes were also expressed in cluster 5 cells (Fig. 2B and D), indicating the multi-potent potential of this cluster. Therefore, our initial annotation of these cells as adipoprimed progenitors was too narrow as it did not include the possible osteogenic differentiation potential. We apologize for the confusion caused by the inappropriate annotation and, in order to avoid any further confusion, cluster 5 has now been re-annotated as ‘highly adipocytic gene-expressing progenitors (HAGEPs), which we believe is a better representation of the cells. We furthermore agree with the reviewer that in-vivo differentiation needs to be performed to address potential differentiation capacities in future studies.
With regard to the lack of adipocytes in our data set, we described in the Materials and Methods section that human bone marrow cells were isolated based on density gradient centrifugation. After centrifugation, the mononuclear cell-containing monolayers were harvested for further analysis. However, the resulting supernatant containing mature adipocytic cells was discarded14. Therefore, adipocyte clusters were not identified in our dataset. We have amended the manuscript accordingly (page 5, line 7).
Regarding the pre-adipocytes, we are not aware of any specific markers for pre-adipocytes in the bone marrow. We examined the only known markers (ICAM1, PPARG, FABP4) that have been shown to mark committed pre-adipocytes in human adipose tissue15. As illustrated in Fig. R1 (below), low expression of all three markers was not restricted to a single distinct cluster but could be found in almost all stromal clusters. These data thus allow us to neither confirm nor exclude the presence of pre-adipocytes in the dataset. Due to the lack of specific markers for pre-adipocytes and the absence of mature adipocytes in the current dataset, it is therefore difficult to identify a well-defined pre-adipocytes cluster.
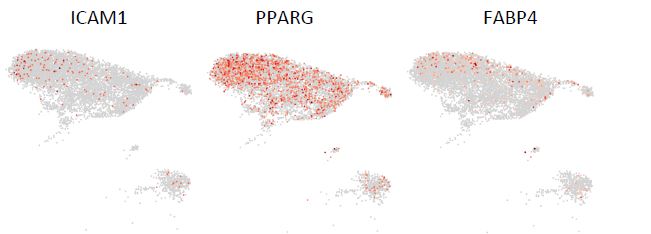
Figure R1. UMAP illustration of the normalized expression of the markers for pre-adipocytes in stromal clusters.
In addition, based on a separate analysis of surface molecules, the authors propose new markers that could be used to prospectively isolate different human subpopulations of BM niche cells by using CD52, CD81 and NCAM1 (=CD56). Indeed, these analyses yield six different populations with differential abilities to form fibroblast-like colonies and differentiate into adipo-, osteo-, and chondrogenic lineages. To explore how the scRNAseq data may help to understand regulatory processes within the BM, the authors predict possible interactions between hematopoietic and non-hematopoietic subpopulations in the BM. These should be further validated, to support statements as the suggestion in the abstract that separate CXCL12- and SPP1-regulated BM niches might exist.
We agree with the reviewer that functional validation of the CellPhoneDB results using for example in vivo humanized mouse models would be needed to demonstrate the presence of different niches in the bone marrow. At this point of time we only put forward the hypothesis that different niche types exist while we will work on providing experimental proof in our future studies.
The scRNAseq analysis is indeed a strong and important resource, also for later studies meant to increase knowledge about the hematopoietic niche of the BM. Although the analyses using different bioinformatic tools is very helpful, they remain mostly speculative, since validatory experiments, as already mentioned, are missing. As such, I feel the authors did not succeed in achieving their goals of understanding how non-hematopoietic cells of the BM regulate the different hematopoietic processes within the BM. Nevertheless, they have created valuable resources, both in the scRNAseq data they generated, as well as the different predictions about different cell populations, their lineage relationships, and how they might interact with hematopoietic cells.
We thank the reviewer for the appreciation of the value of this dataset. We agree with the reviewer that it is of great importance to validate the contribution of potential driver genes for stromal cell differentiation and verify the in vitro data and in-silico prediction using in-vivo models. As the main goal of the current study was to formulate hypotheses based on the scRNAseq data for future studies, we believe that in vivo validation experiments using engineered human bone marrow models or humanized bone marrow ossicles are out of the scope of the current study, but certainly need to be performed in the future.
The impact of this work is difficult to envision, since validations still need to be performed. Also, it has the born in mind that humans are not mice, which can be studied in neat homogeneous inbred populations. Human populations on the other hand, are quite diverse, so that the data generated in this manuscript and others will probably have to be combined to extrapolate data relevant to the whole of the human population. However, as it is equally difficult to generate reliable scRNAseq data from human BM, it seems likely that the data will indeed an important resource, when more data from different donors become available.
We thank the reviewer for the generally positive evaluation of this study.
Taken at point value, the authors provide evidence that human counterparts exist to several BM populations described in mice. In my opinion, the lineage relationships predicted using the RNA velocity analyses need more substance, as it seems the differentiation-paths may diverge from what is known from mice. If so, this issue should be studied more stringently. Similarly, the paper would have been strengthened considerably if a relevant experimental validation would have been attempted, perhaps by using genetically modified (knockdown) MSSC, similar to Battula et al. (doi: 10.1182/blood-2012-06-437988).
In the study from Welner’s group, stromal differentiation trajectory was inferred based on scRNAseq analysis of murine bone marrow cells using Velocyto16. Velocyto identified MSCs as the ‘source’ cell state with pre-adipocytes, pro-osteoblasts, and prochondrocytes being end states. In our study, the MSSC population was predicted to be at the apex of the trajectory and the pre-osteoblast cluster was placed close to the terminal state of differentiation, which is consistent with the murine study. However, different stromal cell types were identified in mice compared with humans. For example, we have identified prefibroblasts in our dataset which are absent in the murine study, while a well-defined murine pre-adipocyte population was not identified in our human dataset. Therefore, it is not surprising to find some discrepancies between human and murine stromal differentiation trajectories. Of course and as mentioned before, critical in-vivo functional validations need to be carried out to address these important issues in the future.
In summary, this is a very interesting but also descriptive paper with highly important resources. However, to prospectively identify or isolate human non-hematopoietic/nonendothelial niche populations, more stringent validations should have been performed to strengthen the validity of the different analyses that have been performed. As such, it remains an open question which niche subpopulations has the most impact on the different hematopoietic processes important for normal and stress hematopoiesis, as well as malignancies.
Thank you for this comment. We completely agree that more stringent validations are necessary but are outside of the aim of our current hypothesis-generating study. Accordingly, we are planning functional verification studies using genetically manipulated stromal cells in combination with in-vivo humanized ossicles. Furthermore, other groups will hopefully use our database and contribute with functional studies in model systems that are currently not available to us, e.g. iPS-derived bone marrow in-vitro proxies.
Specific remarks
• Since CD45, CD235a, and CD271 are used as distinguishing markers in the sample preparation of the scRNAseq, it would be helpful to highlight these markers in the different analyses (Figures 1D, 2B, 2C-F, and 4A), and restrict the analyses to those cells that also not express CD45, CD235a (why use CD71?) and highly express CD271.
Thank you for this comment. As shown in Fig. R2, we have modified figures Fig. 1D, 2B, and 4A showing now also the expression of PTPRC (CD45), GYPA (CD235a), and NGFR (CD271) on the top (Fig. 1D and 2B) or right (Fig. 4A) panel of the figures. To complement Fig. 2C-F, we have generated new stacked violin plots showing the expression level of three markers by all 9 stromal clusters (Fig. R2B). As we believe that including these three markers in the figures does not provide a better strategy to improve the analyses, we decided to leave the original figures unchanged in this respect.
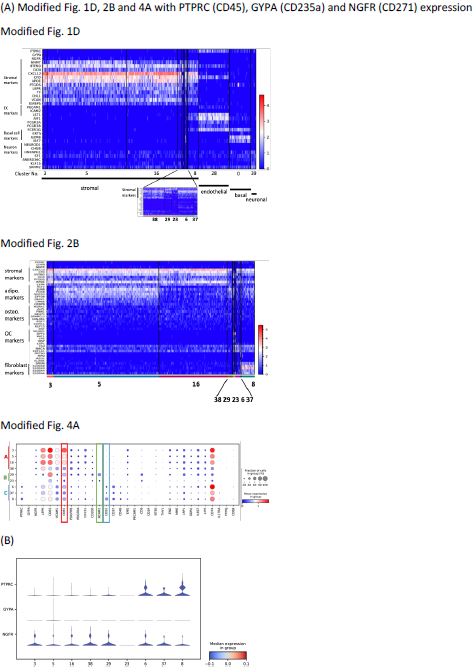
Figure R2. (A) Modified Fig. 1D, 2B and 4A with PTPRC (CD45), GYPA (CD235a) and NGFR (CD271) expression. (B) Stacked violin plots of PTPRC, GYPA and NGFR expressed by stromal clusters to complement Fig. 2C-F.
With regard to cell exclusion based on CD45, as shown in the modified Figure corresponding to Fig 1A in the manuscript (Fig R2A), CD45 gene expression is observed also in the endothelial cluster, basal cluster, and neuronal cluster (Fig. R2A). These clusters represent non-hematopoietic clusters that we would like to keep in our dataset for further analysis, such as cell-cell interaction. Therefore, we choose to not restrict the analysis to solely CD45 nonexpressing cells.
With regard to CD235a (GYPA), expression of CD235a is not detected in any of the nonhematopoietic clusters. Thus, CD235a-expressing cell exclusion is not necessary.
For CD271, according to our previous results (own unpublished data, belonging to a dataset of which only significantly expressed genes were reported in Li et al.8), protein expression of CD271 is not necessarily reflected by gene expression. In the other words, stromal cells with CD271 protein expression do not always have high mRNA expression. A significant fraction of stromal cells would be excluded if we restrict the analyses only to those cells that show high CD271 gene expression, which would not reflect the real cellular composition of human bone marrow stroma. In order to not risk losing stromal cells, we therefore kept our previous analyses which included stromal cells with various CD271 expression levels.
With regard to using CD71 as an exclusion marker, please see also the comments to reviewer 1. Briefly, according to our data, CD71 (TFRC)-expressing erythroid precursors could still be found after excluding CD45 and CD235a positive cells (Figure 1-figure supplement 1B and R3). As furthermore shown in Figure 1-figure supplement 1G and R2, CD71 expression in the stromal clusters is negligible. Therefore, we believe that this justifies the use of CD71 as an additional marker to exclude erythroid cells. We have amended the discussion to address this issue (page 19, lines 7-8).
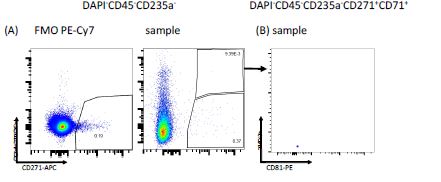
Figure R3. FACS plots illustrating the expression of (A) CD71 (TFRC) vs CD271 in CD45- CD235a- cells and (B) FSC-A vs CD81 in CD45-CD235a-CD271+CD71+ cells following exclusion of doublets and dead cells.
• Despite a distinct neuronal cluster (39), there does not seem to be a distinctive marker for these cells. Is this true?
Yes, the reviewer is correct that there is no significantly-expressed distinctive marker for neuronal cells. Multiple markers indicating the presence of different cell types were identified in cluster 39 (Supplementary File 4). Among them, several neuronal markers (NEUROD1, CHGB, ELAVL2, ELAVL3, ELAVL4, STMN2, INSM1, ZIC2, NNAT) were found to be enriched in this cluster (Supplementary File 4 and Fig. 1D) with higher fold changes compared to other identified genes. However, the expression of these genes was not statistically significant, which is mainly due to the heterogeneity of the cluster and thus does not allow us to draw any firm conclusions.
Several genes including MALAT1, HNRNPH1, AC010970.1, and AD000090.1 were identified to be statistically highly expressed by cluster 39 (Supplementary File 4). The expression of these genes is not restricted to any specific cell type. It is therefore impossible to annotate the cluster based on this and our data thus indicated that cluster 39 is a heterogeneous population containing multiple cell types. Based on the expression of neuronal markers, we nevertheless chose to annotate Cluster 39 as “neuronal” as the prominent expression of neuronal markers indicated the presence of neurons in this cluster. To be more accurate, the annotation of cluster 39 has been changed to ‘neuronal cell-containing cluster’ to correctly reflect the presence of non-neuronal gene expressing cells as well (page 29, lines 3-8).
• Since based on 2C and 2D, the authors are unable to distinguish adipo- from osteogenic cells, would the authors use the same molecules to distinguish different populations of 2C-D, or would they use other markers, if so which and why.
We agree with the reviewer that at the first glance adipo-primed (cluster 5, now annotated as “highly adipocytic gene-expressing progenitors”, HAGEPs), balanced progenitors (cluster 16), and pre-osteoblasts (cluster 38) shared a similar expression pattern according to the violin plots in Fig. 2C and 2D. However, as illustrated in the heatmap (Fig. 2B), the expression patterns of adipo-primed (HAGEP) and balanced progenitors were quite different in terms of their expression of adipogenic and osteogenic markers. Both adipogenic and osteogenic marker expression was detected in HAGEPs, balanced progenitors, and preosteoblasts. Thus, as violin plots are summarizing the overall expression levels of a certain marker in a certain cluster, these plots tend to make it more difficult to detect differential expression patterns between different clusters. In this case, the heatmap shown in Fig. 2B is a good complement to the violin plots as it is demonstrating the different expression patterns of every cell in the different stromal clusters.
Additionally, cluster 5 showed the expression of a group of stress-related transcription factors (FOS, FOSB, JUNB, EGR1) (Fig. 2B and Figure 2-figure supplement 1C), some of which had been shown to mark bone marrow adipogenic progenitors1. The expression of the abovementioned stress-related transcription factors (putative adipogenic progenitor markers) was generally lower in cluster 38 compared to cluster 5, further demonstrating that clusters were different.
Furthermore, there was a gradual upregulation of more mature osteogenic markers such as RUNX1, CDH11, EBF1, and EBF3 from cluster 5 to cluster 16 and finally cluster 38. As shown in Fig. 2D, the expression of these markers was higher in cluster 38 compared to cluster 5. Therefore, cluster 38 was annotated as pre-osteoblasts.
Most of the stromal clusters form a continuum (Fig. 2A), which correlates very well with the gradual transition of different cellular states during stromal cell development. It is highly unlikely that abrupt and dramatic gene expression changes would occur during the cellular state transition of cells of the same lineage. Therefore, it is not surprising to find the differences in gene expression profiles between stromal clusters share a certain level of similarities.
In summary, we rely on several factors to distinguish different stromal clusters, which include canonical adipo-, osteo- and chondrogenic markers, stress markers, heatmap, violin plots, and the gradual up-regulation of certain lineage-specific markers.
To directly answer the reviewer’s question, we believe that we are able to distinguish different stromal clusters based on our data.
• In de Jong et al., an inflammatory MSC population (iMSC) is defined. Since the Schneider group showed that inflammatory S100A8 and A9 are expressed by inflamed MSC, is it possible that the some of the designated pre-fibroblasts actually correspond to these S100A8/A9-expressing iMSC?
We thank the reviewer for raising this interesting question.
First of all, we would like to point out that scRNAseq was performed using viably frozen bone marrow aspirates in de Jong’s study while freshly isolated bone marrows were used in our study. There might be discrepancies between frozen and fresh bone marrow samples in terms of cellular composition including stromal composition and, importantly, processinginduced stress-related gene expression profiles.
To investigate if designated pre-fibroblasts actually correspond to iMSCs as suggested by the reviewer, we have re-examined the expression of some of the key iMSC genes as reported by de Jong et al 17. As shown in Fig. R6, the markers that can distinguish iMSC from other MSC clusters in de Jong et al. study were not exclusively expressed by pre-fibroblasts, but also by other stromal cell types including HAGEPs, balanced progenitors, and pre-osteoblasts.
In the study by R. Schneider’s group18, significant upregulation of S100A8/S100A9 was observed in stromal cells from patients with myelofibrosis. Furthermore, base-line expression of S100A8/A9 was also observed in the fibroblast clusters in the control group, which correlates very well with our data of S100A8/9 expression in pre-fibroblasts in normal donors (Fig. 2F). Our data thus indicate – in line with Schneider’s findings - that there is a baseline level expression of S100A8/9 in fibroblasts in hematologically normal samples and that the expression of S100A8/9 is not restricted to inflamed MSC.
In summary, the gene expression profiles observed in our study do not indicate the presence of iMSC in the healthy bone marrow.
• Figure 3A: Do human adipo-primed cells (cluster 5) indeed differentiate into osteogenic cells (clusters 6, 38, and 39). This would be highly unexpected. Can the authors substantiate this "reliable outcome of the RNA velocity analysis"?
Please refer to our previous responses regarding this topic. Briefly, as shown in Fig. 2B and D, both osteogenic and adipogenic genes are expressed in cluster 5, indicating the multi-potent potentials of this cluster. Although the cluster was initially annotated as adipo-primed progenitors, this was not intended to exclude the osteogenic differentiation potential of these progenitors. Nevertheless, this annotation did not correctly reflect the differentiation potential and might thus have caused confusion, for which we apologize. In order to more correctly describe the characteristics of these cells, cluster 5 has now been reannotated as ‘highly adipocytic gene-expressing progenitors (HAGEPs)’.
In general, the outcome of the RNA velocity analysis needs to be corroborated by in-vivo differentiation experiments. But we believe that functional verification, which would be extensive, is out of the scope of the current study and we will address these questions in future studies.
• How statistically certain are the authors, that the populations in Figure 4B as defined by flow cytometry, correspond to MSSC, adipo-primed cells, osteoprogenitors, etc., as defined by scRNAseq?
To address this question, we sorted the A1-A4 populations and performed RT- PCR to examine the CD81 expression level in each cluster. As shown in Figure 4-figure supplement 1B, CD81 expression levels were higher in A1 and A2 compared with A3 and A4, which is consistent with the scRNAseq data that showed the highest CD81 expression in MSSCs compared to other clusters (Supplementary File 4).
The phenotypes defined in this study allowed us to isolate different stromal cell types which demonstrated significant functional differences as described in the manuscript (page 19, lines 17-25; page 20, lines 1-11). These results, in combination with the quantitative real-time PCR results (Figure 4-figure supplement 1B), demonstrated that the A1-A4 subsets in FACS are functionally distinct populations and are likely to be – at least in large parts – identical or equivalent to the transcriptionally identified clusters in group A stromal cells. However, at this point, we do not have performed the required experiments (scRNAseq of sorted cells) that would provide sufficient proof to confirm this statement statistically.
• The immunohistochemistry results shown do not allow distinct conclusions as the colors give unequivocal mix-colors, and surface expression cannot be distinguished from intracellular expression. Please use a 3D (confocal) method for such statements.
We thank the reviewer for the suggestion and we have performed additional confocal microscopy analysis of human bone marrow biopsies as suggested by the reviewer. Representative confocal images are now presented in the middle and right panel of Fig. 6E. We also include a separate file (Supplemental confocal image file). Here, confocal scans of all maker combinations are shown as ortho views in addition to detailed intensity profile analyses of the cells of interest clearly distinguishing surface staining from intracellular staining.
Confocal analysis of bone marrow biopsies confirmed our findings presented in the manuscript. As observed in the scanning images, CD271-expressing cells were negative for CD45 and were located in perivascular, endosteal, and peri-adipocytic regions. CD271/CD81double positive cells could be found either in the peri-adipocytic regions or perivascular regions while CD271/NCAM1 double-positive cells were exclusively situated at the bone-lining endosteal regions. The results of the confocal analysis have been added to the revised manuscript (page 21, lines 15-17).
• Figure 5A: as all cells seem to interact with all other cells, this figure does not convey relevant information about BM regions using for instance CXCL12 or SPP1. Please reanalyze to show specificity of the interactions of the single clusters. Also, since it is unlikely the CellPhoneDB2-predicted interactions are restricted to hematopoietic responders, please also describe the possible interactions between non-hematopoietic cells.
Fig. 5A was used to demonstrate the complexity of the interactions between hematopoietic cells and stromal cells.
To gain a more detailed understanding of the interactions, we also performed an analysis with the top-listed ligand-receptor pairs as shown in Fig. 5B-C and Figure 5-figure supplement 1B. Here, each dot represents the interaction of a specific ligand-receptor pair listed on the x-axis between the two individual clusters indicated in the y-axis, which we believe shows what the reviewer is asking for.
The specificity of the interactions between single clusters were shown in Fig. 5B-C and Figure 5-figure supplement 1B. The CXCL12- and SPP1-mediated interactions between MSSC/OC and hematopoietic clusters clearly suggested stromal cell type-specific interactions.
Regarding non-hematopoietic cells, both inter- and intra-stromal interactions were identified to be operative between different stromal subsets as well as within the same stromal cell population as shown in Figure 5-figure supplement 3B. In addition, we have also analyzed the interaction pattern between endothelial cells and hematopoietic cells as shown in Fig. 7A, and thus we believe that we have sufficiently described these interactions as requested by the reviewer.
-
Evaluation Summary:
The manuscript by Li and coworkers characterizes sorted human non-hematopoietic bone marrow cells by scRNA-seq and predicts their lineage relationships and possible interactions with mature and immature hematopoietic cells. Transcriptionally-different stromal cell subsets are identified, and their lineage relationships, cell-cell interactions and possible specialized functions are inferred or predicted from in-silico studies, paving the way for future functional and validation studies. This resource significantly adds to the current understanding human non-hematopoietic bone marrow stromal cells and their hematopoietic regulatory functions.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the …
Evaluation Summary:
The manuscript by Li and coworkers characterizes sorted human non-hematopoietic bone marrow cells by scRNA-seq and predicts their lineage relationships and possible interactions with mature and immature hematopoietic cells. Transcriptionally-different stromal cell subsets are identified, and their lineage relationships, cell-cell interactions and possible specialized functions are inferred or predicted from in-silico studies, paving the way for future functional and validation studies. This resource significantly adds to the current understanding human non-hematopoietic bone marrow stromal cells and their hematopoietic regulatory functions.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 and Reviewer #2 agreed to share their name with the authors.)
-
Reviewer #1 (Public Review):
This is an awesome comprehensive manuscript. Authors start by sorting putative stromal cell-containing BM non-hematopoietic (CD235a-/CD45-) plus additional CD271+/CD235a-/CD45- populations to identify nine individual stromal identities by scRNA-seq. The dual sorting strategy is a clever trick as it enriches for rare stromal (progenitor) cell signals but may suffer a certain bias towards CD271+ stromal progenitors. The lack of readable signatures already among CD45-/CD45- sorts might argue against this fear. This reviewer would appreciate a brief discussion on number & phenotype of putative additional MSSC phenotypes in light of the fact that the majority of 'blood lineage(s)'-negative scRNA-seq signatures identified blood cell progenitor identities (glycophorin A-negative & leukocyte common …
Reviewer #1 (Public Review):
This is an awesome comprehensive manuscript. Authors start by sorting putative stromal cell-containing BM non-hematopoietic (CD235a-/CD45-) plus additional CD271+/CD235a-/CD45- populations to identify nine individual stromal identities by scRNA-seq. The dual sorting strategy is a clever trick as it enriches for rare stromal (progenitor) cell signals but may suffer a certain bias towards CD271+ stromal progenitors. The lack of readable signatures already among CD45-/CD45- sorts might argue against this fear. This reviewer would appreciate a brief discussion on number & phenotype of putative additional MSSC phenotypes in light of the fact that the majority of 'blood lineage(s)'-negative scRNA-seq signatures identified blood cell progenitor identities (glycophorin A-negative & leukocyte common antigen-negative). The nine stromal cell entities share the CXCL12, VCAN, LEPR main signature. Perhaps the authors could speculate if future studies using VCAN or LEPR-based sort strategies could identify additional stromal progenitor identities?
The authors furthermore localized CD271+, CD81+ and NCAM/CD56+ cells in BM sections in situ. Finally, referring to the strong background of the group in HSC research, in silico prediction by CellPhoneDB identified a wide range of interactions between stromal cells and hematopoietic cells. Evidence for functional interdependence of FCU-F forming cells is completing the novel and more clear bone marrow stromal cell picture.
An illustrative abstract naming the top9 stromal identities in their top4 clusters by their "top10 markers" + functions would be highly appreciated. -
Reviewer #2 (Public Review):
Knowledge about composition and function of the different subpopulations of the hematopoietic niche of the BM is limited. Although such knowledge about the mouse BM has been accumulating in recent years, a thorough study of the human BM still needs to be performed. The present manuscript of Li and coworkers fills this gap by performing single cell RNA sequencing (scRNAseq) on control BM as well as CD271+ BM cells enriched for non-hematopoietic niche cells.
Based on their scRNAseq, the authors propose 41 different BM cell populations, ten of which represented non-hematopoietic cells, including one endothelial cell cluster. The nine remaining skeletal subpopulations were subdivided into multipotent stromal stem cells (MSSC), four distinct populations of osteoprogenitors, one cluster of osteoblasts and three …
Reviewer #2 (Public Review):
Knowledge about composition and function of the different subpopulations of the hematopoietic niche of the BM is limited. Although such knowledge about the mouse BM has been accumulating in recent years, a thorough study of the human BM still needs to be performed. The present manuscript of Li and coworkers fills this gap by performing single cell RNA sequencing (scRNAseq) on control BM as well as CD271+ BM cells enriched for non-hematopoietic niche cells.
Based on their scRNAseq, the authors propose 41 different BM cell populations, ten of which represented non-hematopoietic cells, including one endothelial cell cluster. The nine remaining skeletal subpopulations were subdivided into multipotent stromal stem cells (MSSC), four distinct populations of osteoprogenitors, one cluster of osteoblasts and three clusters of pre-fibroblasts. Using bioinformatic tools, the authors then compare their results and divisions of subpopulations to some previously published work from others and attempt to delineate lineage relationships using RNA velocity analyses. From these, they propose different paths from which MSSC enter the progenitor stages, and might differentiate into pre-osteoblasts and -fibroblasts.
It is of interest to note, that apparently adipo-primed cells may also differentiate into osteolineage cells, something that should be further explored or validated. Furthermore, although this analysis yields a large adipo-primed populations, pre-adipocytes and mature adipocytes appear not to be included in the data set the authors used, which should also be explained.
In addition, based on a separate analysis of surface molecules, the authors propose new markers that could be used to prospectively isolate different human subpopulations of BM niche cells by using CD52, CD81 and NCAM1 (=CD56). Indeed, these analyses yield six different populations with differential abilities to form fibroblast-like colonies and differentiate into adipo-, osteo-, and chondrogenic lineages. To explore how the scRNAseq data may help to understand regulatory processes within the BM, the authors predict possible interactions between hematopoietic and non-hematopoietic subpopulations in the BM. These should be further validated, to support statements as the suggestion in the abstract that separate CXCL12- and SPP1-regulated BM niches might exist.
The scRNAseq analysis is indeed a strong and important resource, also for later studies meant to increase knowledge about the hematopoietic niche of the BM. Although the analyses using different bioinformatic tools is very helpful, they remain mostly speculative, since validatory experiments, as already mentioned, are missing. As such, I feel the authors did not succeed in achieving their goals of understanding how non-hematopoietic cells of the BM regulate the different hematopoietic processes within the BM. Nevertheless, they have created valuable resources, both in the scRNAseq data they generated, as well as the different predictions about different cell populations, their lineage relationships, and how they might interact with hematopoietic cells.
The impact of this work is difficult to envision, since validations still need to be performed. Also, it has the born in mind that humans are not mice, which can be studied in neat homogeneous inbred populations. Human populations on the other hand, are quite diverse, so that the data generated in this manuscript and others will probably have to be combined to extrapolate data relevant to the whole of the human population. However, as it is equally difficult to generate reliable scRNAseq data from human BM, it seems likely that the data will indeed an important resource, when more data from different donors become available.
Taken at point value, the authors provide evidence that human counterparts exist to several BM populations described in mice. In my opinion, the lineage relationships predicted using the RNA velocity analyses need more substance, as it seems the differentiation-paths may diverge from what is known from mice. If so, this issue should be studied more stringently. Similarly, the paper would have been strengthened considerably if a relevant experimental validation would have been attempted, perhaps by using genetically modified (knockdown) MSSC, similar to Battula et al. (doi: 10.1182/blood-2012-06-437988).
In summary, this is a very interesting but also descriptive paper with highly important resources. However, to prospectively identify or isolate human non-hematopoietic/non-endothelial niche populations, more stringent validations should have been performed to strengthen the validity of the different analyses that have been performed. As such, it remains an open question which niche subpopulations has the most impact on the different hematopoietic processes important for normal and stress hematopoiesis, as well as malignancies.
Specific remarks
• Since CD45, CD235a, and CD271 are used as distinguishing markers in the sample preparation of the scRNAseq, it would be helpful to highlight these markers in the different analyses (Figures 1D, 2B, 2C-F, and 4A), and restrict the analyses to those cells that also not express CD45, CD235a (why use CD71?) and highly express CD271.
• Despite a distinct neuronal cluster (39), there does not seem to be a distinctive marker for these cells. Is this true?
• Since based on 2C and 2D, the authors are unable to distinguish adipo- from osteogenic cells, would the authors use the same molecules to distinguish different populations of 2C-D, or would they use other markers, if so which and why.
• In de Jong et al., an inflammatory MSC population (iMSC) is defined. Since the Schneider group showed that inflammatory S100A8 and A9 are expressed by inflamed MSC, is it possible that the some of the designated pre-fibroblasts actually correspond to these S100A8/A9-expressing iMSC?
• Figure 3A: Do human adipo-primed cells (cluster 5) indeed differentiate into osteogenic cells (clusters 6, 38, and 39). This would be highly unexpected. Can the authors substantiate this "reliable outcome of the RNA velocity analysis"?
• How statistically certain are the authors, that the populations in Figure 4B as defined by flow cytometry, correspond to MSSC, adipo-primed cells, osteoprogenitors, etc., as defined by scRNAseq?
• The immunohistochemistry results shown do not allow distinct conclusions as the colors give unequivocal mix-colors, and surface expression cannot be distinguished from intracellular expression. Please use a 3D (confocal) method for such statements.
• Figure 5A: as all cells seem to interact with all other cells, this figure does not convey relevant information about BM regions using for instance CXCL12 or SPP1. Please re-analyze to show specificity of the interactions of the single clusters. Also, since it is unlikely the CellPhoneDB2-predicted interactions are restricted to hematopoietic responders, please also describe the possible interactions between non-hematopoietic cells. -
