A unified view of low complexity regions (LCRs) across species
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
This paper describes a new dotplot-based approach for analyzing Low Complexity Regions (LCRs) in proteins. The work is validated against a single protein, compared to existing methods, and applied to the proteomes of several model systems. The work aims to show links between specific LCRs and biological function and subcellular location, and study conservation in LCRs amongst higher species.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #2 agreed to share their name with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Low complexity regions (LCRs) play a role in a variety of important biological processes, yet we lack a unified view of their sequences, features, relationships, and functions. Here, we use dotplots and dimensionality reduction to systematically define LCR type/copy relationships and create a map of LCR sequence space capable of integrating LCR features and functions. By defining LCR relationships across the proteome, we provide insight into how LCR type and copy number contribute to higher order assemblies, such as the importance of K-rich LCR copy number for assembly of the nucleolar protein RPA43 in vivo and in vitro. With LCR maps, we reveal the underlying structure of LCR sequence space, and relate differential occupancy in this space to the conservation and emergence of higher order assemblies, including the metazoan extracellular matrix and plant cell wall. Together, LCR relationships and maps uncover and identify scaffold-client relationships among E-rich LCR-containing proteins in the nucleolus, and revealed previously undescribed regions of LCR sequence space with signatures of higher order assemblies, including a teleost-specific T/H-rich sequence space. Thus, this unified view of LCRs enables discovery of how LCRs encode higher order assemblies of organisms.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
In this manuscript, the authors present a new technique for analysing low complexity regions (LCRs) in proteins- extended stretches of amino acids made up from a small number of distinct residue types. They validate their new approach against a single protein, compare this technique to existing methods, and go on to apply this to the proteomes of several model systems. In this work, they aim to show links between specific LCRs and biological function and subcellular location, and then study conservation in LCRs amongst higher species.
The new method presented is straightforward and clearly described, generating comparable results with existing techniques. The technique can be easily applied to new problems and the authors have made code available.
This paper is less successful in …
Author Response
Reviewer #1 (Public Review):
In this manuscript, the authors present a new technique for analysing low complexity regions (LCRs) in proteins- extended stretches of amino acids made up from a small number of distinct residue types. They validate their new approach against a single protein, compare this technique to existing methods, and go on to apply this to the proteomes of several model systems. In this work, they aim to show links between specific LCRs and biological function and subcellular location, and then study conservation in LCRs amongst higher species.
The new method presented is straightforward and clearly described, generating comparable results with existing techniques. The technique can be easily applied to new problems and the authors have made code available.
This paper is less successful in drawing links between their results and the importance biologically. The introduction does not clearly position this work in the context of previous literature, using relatively specialised technical terms without defining them, and leaving the reader unclear about how the results have advanced the field. In terms of their results, the authors further propose interesting links between LCRs and function. However, their analyses for these most exciting results rely heavily on UMAP visualisation and the use of tests with apparently small effect sizes. This is a weakness throughout the paper and reduces the support for strong conclusions.
We appreciate the reviewer’s comments on our manuscript. To address comments about the clarity of the introduction and the position of our findings with respect to the rest of the field, we have made several changes to the text. We have reworked the introduction to provide a clearer view of the current state of the LCR field, and our goals for this manuscript. We also have made several changes to the beginnings and ends of several sections in the Results to explicitly state how each section and its findings help advance the goal we describe in the introduction, and the field more generally. We hope that these changes help make the flow of the paper more clear to the reader, and provide a clear connection between our work and the field.
We address comments about the use of UMAPs and statistical tests in our responses to the specific comments below.
Additionally, whilst the experimental work is interesting and concerns LCRs, it does not clearly fit into the rest of the body of work focused as it is on a single protein and the importance of its LCRs. It arguably serves as a validation of the method, but if that is the author's intention it needs to be made more clearly as it appears orthogonal to the overall drive of the paper.
In response to this comment, we have made more explicit the rationale for choosing this protein at the beginning of this section, and clarify the role that these experiments play in the overall flow of the paper.
Our intention with the experiments in Figure 2 was to highlight the utility of our approach in understanding how LCR type and copy number influence protein function. Understanding how LCR type and copy number can influence protein function is clearly outlined as a goal of the paper in the Introduction.
In the text corresponding to Figure 2, we hypothesize how different LCR relationships may inform the function of the proteins that have them, and how each group in Figure 2A/B can be used to test these hypotheses. The global view provided by our method allows proteins to be selected on the basis of their LCR type and copy number for further study.
To demonstrate the utility of this view, we select a key nucleolar protein with multiple copies of the same LCR type (RPA43, a subunit of RNA Pol I), and learn important features driving its higher-order assembly in vivo and in vitro. We learned that in vivo, a least two copies of RPA43’s K-rich LCRs are required for nucleolar integration, and that these K-rich LCRs are also necessary for in vitro phase separation.
Despite this protein being a single example, we were able to gain important insights about how K-rich LCR copy number affects protein function, and that both in vitro higher order assembly and in vivo nucleolar integration can be explained by LCR copy number. We believe this opens the door to ask further questions about LCR type and copy number for other proteins using this line of reasoning.
Overall I think the ideas presented in the work are interesting, the method is sound, but the data does not clearly support the drawing of strong conclusions. The weakness in the conclusions and the poor description of the wider background lead me to question the impact of this work on the broader field.
For all the points where Reviewer #1 comments on the data and its conclusions, we provide explanations and additional analyses in our responses below showing that the data do indeed support our conclusions. In regards to our description of the wider background, we have reworked our introduction to more clearly link our work to the broader field, such that a more general audience can appreciate the impact of our work.
Technical weaknesses
In the testing of the dotplot based method, the manuscript presents a FDR rate based on a comparison between real proteome data and a null proteome. This is a sensible approach, but their choice of a uniform random distribution would be expected to mislead. This is because if the distribution is non-uniform, stretches of the most frequent amino will occur more frequently than in the uniform distribution.
Thank you for pointing this out. The choice of null proteome was a topic of much discussion between the authors as this work was being performed. While we maintain that the uniform background is the most appropriate, the question from this reviewer and the other reviewers made us realize that a thorough explanation was warranted. For a complete explanation for our choice of this uniform null model, please see the newly added appendix section, Appendix 1.
The authors would also like to point out that the original SEG algorithm (Wootton and Federhen, 1993) also made the intentional choice of using a uniform background model.
More generally I think the results presented suggest that the results dotplot generates are comparable to existing methods, not better and the text would be more accurate if this conclusion was clearer, in the absence of an additional set of data that could be used as a "ground truth".
We did not intend to make any strong claims about the relative performance of our approach vs. existing methods with regard to the sequence entropy of the called LCRs beyond them being comparable, as this was not the main focus of our paper. To clarify the text such that it reflects this, we have removed ‘or better’ from the text in this section.
The authors draw links between protein localisation/function and LCR content. This is done through the use of UMAP visualisation and wilcoxon rank sum tests on the amino acid frequency in different localisations. This is convincing in the case of ECM data, but the arguments are substantially less clear for other localisations/functions. The UMAP graphics show generally that the specific functions are sparsely spread. Moreover when considering the sample size (in the context of the whole proteome) the p-value threshold obscures what appear to be relatively small effect sizes.
We would first like to note that some of the amino acid frequency biases have been documented and experimentally validated by other groups, as we write and reference in the manuscript. Nonetheless, we have considered the reviewer's concerns, and upon rereading the section corresponding to Figure 3, we realize that our wording may have caused confusion in the interpretation there. In addition to clarifying this in the manuscript, we believe the following clarification may help in the interpretations drawn from that section.
Each point in this analysis (and on the UMAP) is an LCR from a protein, and as such multiple LCRs from the same protein will appear as multiple points. This is particularly relevant for considering the interpretation of the functional/higher order assembly annotations because it is not expected that for a given protein, all of the LCRs will be directly relevant to the function/annotation. Just because proteins of an assembly are enriched for a given type of LCR does not mean that they only have that kind of LCR. In addition to the enriched LCR, they may or may not have other LCRs that play other roles.
For example, a protein in the Nuclear Speckle may contain both an R/S-rich LCR and a Q-rich LCR. When looking at the Speckle, all of the LCRs of a protein are assigned this annotation, and so such a protein would contribute a point in the R/S region as well as elsewhere on the map. Because such "non-enriched" LCRs do not occur as frequently, and may not be relevant to Speckle function, they are sparsely spread.
We have now changed the wording in that section of the main text to reflect that the expectation is not all LCRs mapping to a certain region, but enrichment of certain LCR compositions.
Reviewer #3 (Public Review):
The authors present a systematic assessment of low complexity sequences (LCRs) apply the dotplot matrix method for sequence comparison to identify low-complexity regions based on per-residue similarity. By taking the resulting self-comparison matrices and leveraging tools from image processing, the authors define LCRs based on similarity or non-similarity to one another. Taking the composition of these LCRs, the authors then compare how distinct regions of LCR sequence space compare across different proteomes.
The paper is well-written and easy to follow, and the results are consistent with prior work. The figures and data are presented in an extremely accessible way and the conclusions seem logical and sound.
My big picture concern stems from one that is perhaps challenging to evaluate, but it is not really clear to me exactly what we learn here. The authors do a fine job of cataloging LCRs, offer a number of anecdotal inferences and observations are made - perhaps this is sufficient in terms of novelty and interest, but if anyone takes a proteome and identifies sequences based on some set of features that sit in the tails of the feature distribution, they can similarly construct intriguing but somewhat speculative hypotheses regarding the possible origins or meaning of those features.
The authors use the lysine-repeats as specific examples where they test a hypothesis, which is good, but the importance of lysine repeats in driving nucleolar localization is well established at this point - i.e. to me at least the bioinformatics analysis that precedes those results is unnecessary to have made the resulting prediction. Similarly, the authors find compositional biases in LCR proteins that are found in certain organelles, but those biases are also already established. These are not strictly criticisms, in that it's good that established patterns are found with this method, but I suppose my concern is that this is a lot of work that perhaps does not really push the needle particularly far.
As an important caveat to this somewhat muted reception, I recognize that having worked on problems in this area for 10+ years I may also be displaying my own biases, and perhaps things that are "already established" warrant repeating with a new approach and a new light. As such, this particular criticism may well be one that can and should be ignored.
We thank the reviewer for taking the time to read and give feedback for our manuscript. We respectfully disagree that our work does not push the needle particularly far.
In the section titled ‘LCR copy number impacts protein function’, our goal is not to highlight the importance of lysines in nucleolar localization, but to provide a specific example of how studying LCR copy number, made possible by our approach, can provide specific biological insights. We first show that K-rich LCRs can mediate in vitro assembly. Moreover, we show that the copy number of K-rich LCRs is important for both higher order assembly in vitro and nucleolar localization in cells, which suggests that by mediating interactions, K-rich LCRs may contribute to the assembly of the nucleolus, and that this is related to nucleolar localization. The ability of our approach to relate previously unrelated roles of K-rich LCRs not only demonstrates the value of a unified view of LCRs but also opens the door to study LCR relationships in any context.
Furthermore, our goal in identifying established biases in LCR composition for certain assemblies was to validate that the sequence space captures higher order assemblies which are known. In addition to known biases, we use our approach to uncover the roles of LCR biases that have not been explored (e.g. E-rich LCRs in nucleoli, see Figure 4 in revised manuscript), and discover new regions of LCR sequence space which have signatures of higher order assemblies (e.g. Teleost-specific T/H-rich LCRs). Collectively, our results show that a unified view of LCRs relates the disparate functions of LCRs.
In response to these comments, we have added additional explanations at the end of several sections to clarify the impact of our findings in the scope of the broader field. Furthermore, as we note in our main response, we have added experimental data with new findings to address this concern.
That overall concern notwithstanding, I had several other questions that sprung to mind.
Dotplot matrix approach
The authors do a fantastic job of explaining this, but I'm left wondering, if one used an algorithm like (say) SEG, defined LCRs, and then compared between LCRs based on composition, would we expect the results to be so different? i.e. the authors make a big deal about the dotplot matrix approach enabling comparison of LCR type, but, it's not clear to me that this is just because it combines a two-step operation into a one-step operation. It would be useful I think to perform a similar analysis as is done later on using SEG and ask if the same UMAP structure appears (and discuss if yes/no).
Thank you for your thoughtful question about the differences between SEG and the dotplot matrix approach. We have tried our best to convey the advantages of the dotplot approach over SEG in the paper, but we did not focus on this for the following reasons:
SEG and dotplot matrices are long-established approaches to assessing LCRs. We did not see it in the scope of our paper to compare between these when our main claim is that the approach as a whole (looking at LCR sequence, relationships, features, and functions) is what gives a broader understanding of LCRs across proteomes. The key benefits of dotplots, such as direct visual interpretation, distinguishing LCR types and copy number within a protein, are conveyed in Figure 1A-C and Figure 1 - figure supplements 1 and 4. In fact, these benefits of dotplots were acknowledged in the early SEG papers, where they recommended using dotplots to gain a prior understanding of protein sequences of interest, when it was not yet computationally feasible to analyze dotplots on the same scale as SEG (Wootton and Federhen, Methods in Enzymology, vol. 266, 1996, Pages 554-571). Thus, our focus is on the ability to utilize image processing tools to "convert" the intuition of dotplots into precise read-out of LCRs and their relationships on a multi-proteome scale. All that being said, we have considered differences between these methods as you can see from our technical considerations in part 2 below.
SEG takes an approach to find LCRs irrespective of the type of LCR, primarily because SEG was originally used to mask LCR-containing regions in proteins to facilitate studies of globular domains. Because of this, the recommended usage of SEG commonly fuses nearby LCRs and designates the entire region as "low complexity". For the original purpose of SEG, this is understandable because it takes a very conservative approach to ensure that the non-low complexity regions (i.e. putative folded domains) are well-annotated. However, for the purpose of distinguishing LCR composition, this is not ideal because it is not stringent in separating LCRs that are close together, but different in composition. Fusion can be seen in the comparison of specific LCR calls of the collagen CO1A1 (Figure 1 - figure supplement 3E), where even the intermediate stringency SEG settings fuse LCR calls that the dotplot approach keeps separate. Finally, we did also try downstream UMAP analysis with LCRs called from SEG, and found that although certain aspects of the dotplot-based LCR UMAP are reflected in the SEG-based LCR UMAP, there is overall worse resolution with default settings, which is likely due to fused LCRs of different compositions. Attempting to improve resolution using more stringent settings comes at the cost of the number of LCRs assessed. We have attached this analysis to our rebuttal for the reviewer, but maintain that this comparison is not really the focus of our manuscript. We do not make strong claims about the dotplot matrices being better at calling LCRs than SEG, or any other method.
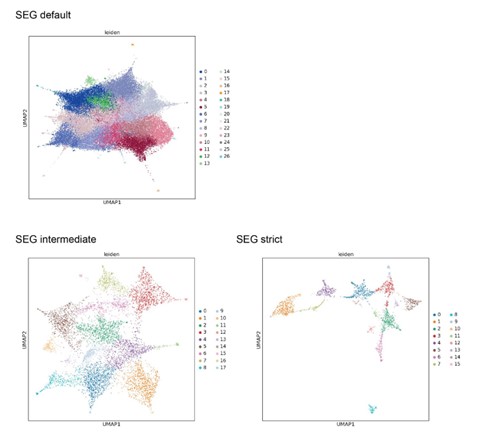
UMAPs generated from LCRs called by SEG
LCRs from repeat expansions
I did not see any discussion on the role that repeat expansions can play in defining LCRs. This seems like an important area that should be considered, especially if we expect certain LCRs to appear more frequently due to a combination of slippy codons and minimal impact due to the biochemical properties of the resulting LCR. The authors pursue a (very reasonable) model in which LCRs are functional and important, but it seems the alternative (that LCRs are simply an unavoidable product of large proteomes and emerge through genetic events that are insufficiently deleterious to be selected against). Some discussion on this would be helpful. it also makes me wonder if the authors' null proteome model is the "right" model, although I would also say developing an accurate and reasonable null model that accounts for repeat expansions is beyond what I would consider the scope of this paper.
While the role of repeat expansions in generating LCRs has been studied and discussed extensively in the LCR field, we decided to focus on the question of which LCRs exist in the proteome, and what may be the function downstream of that. The rationale for this is that while one might not expect a functional LCR to arise from repeat expansion, this argument is less of a concern in the presence of evidence that these LCRs are functional. For example, for many of these LCRs (e.g. a K-rich LCR, R/S-rich LCR, etc as in Figure 3), we know that it is sufficient for the integration of that sequence into the higher order assembly. Moreover, in more recent cases, variation of the length of an LCR was shown to have functional consequences (Basu et al., Cell, 2020), suggesting that LCR emergence through repeat expansions does not imply lack of function. Therefore, while we think the origin of a LCR is an interesting question, whether or not that LCR was gained through repeat expansions does not fall into the scope of this paper.
In regards to repeat expansions as it pertains to our choice of null model, we reasoned that because the origin of an LCR is not necessarily coupled to its function, it would be more useful to retain LCR sequences even if they may be more likely to occur given a background proteome composition. This way, instead of being tossed based on an assumption, LCRs can be evaluated on their function through other approaches which do not assume that likelihood of occurrence inversely relates to function.
While we maintain that the uniform background is the most appropriate, the question from this reviewer and the other reviewers made us realize that a thorough explanation was warranted for this choice of null proteome. For a complete explanation for our choice of this uniform null model, please see the newly added appendix section, Appendix 1.
The authors would also like to point out that the original SEG algorithm (Wootton and Federhen, 1993) also made the intentional choice of using a uniform background model.
Minor points
Early on the authors discuss the roles of LCRs in higher-order assemblies. They then make reference to the lysine tracts as having a valence of 2 or 3. It is possibly useful to mention that valence reflects the number of simultaneous partners that a protein can interact with - while it is certainly possible that a single lysine tracts interacts with a single partner simultaneously (meaning the tract contributes a valence of 1) I don't think the authors can know that, so it may be wise to avoid specifying the specific valence.
Thank you for pointing this out. We agree with the reviewer's interpretation and have removed our initial interpretation from the text and simply state that a copy number of at least two is required for RPA43’s integration into the nucleolus.
The authors make reference to Q/H LCRs. Recent work from Gutiérrez et al. eLife (2022) has argued that histidine-richness in some glutamine-rich LCRs is above the number expected based on codon bias, and may reflect a mode of pH sensing. This may be worth discussing.
We appreciate the reviewer pointing out this publication. While this manuscript wasn’t published when we wrote our paper, upon reading it we agree it has some very relevant findings. We have added a reference to this manuscript in our discussion when discussing Q/H-rich LCRs.
Eric Ross has a number of very nice papers on this topic, but sadly I don't think any of them are cited here. On the question of LCR composition and condensate recruitment, I would recommend Boncella et al. PNAS (2020). On the question of proteome-wide LCR analysis, see Cascarina et al PLoS CompBio (2018) and Cascarina et al PLoS CompBio 2020.
We appreciate the reviewer for noting this related body of work. We have updated the citations to include work from Eric Ross where relevant.
-
Evaluation Summary:
This paper describes a new dotplot-based approach for analyzing Low Complexity Regions (LCRs) in proteins. The work is validated against a single protein, compared to existing methods, and applied to the proteomes of several model systems. The work aims to show links between specific LCRs and biological function and subcellular location, and study conservation in LCRs amongst higher species.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #2 agreed to share their name with the authors.)
-
Reviewer #1 (Public Review):
In this manuscript, the authors present a new technique for analysing low complexity regions (LCRs) in proteins- extended stretches of amino acids made up from a small number of distinct residue types. They validate their new approach against a single protein, compare this technique to existing methods, and go on to apply this to the proteomes of several model systems. In this work, they aim to show links between specific LCRs and biological function and subcellular location, and then study conservation in LCRs amongst higher species.
The new method presented is straightforward and clearly described, generating comparable results with existing techniques. The technique can be easily applied to new problems and the authors have made code available.
This paper is less successful in drawing links between their …
Reviewer #1 (Public Review):
In this manuscript, the authors present a new technique for analysing low complexity regions (LCRs) in proteins- extended stretches of amino acids made up from a small number of distinct residue types. They validate their new approach against a single protein, compare this technique to existing methods, and go on to apply this to the proteomes of several model systems. In this work, they aim to show links between specific LCRs and biological function and subcellular location, and then study conservation in LCRs amongst higher species.
The new method presented is straightforward and clearly described, generating comparable results with existing techniques. The technique can be easily applied to new problems and the authors have made code available.
This paper is less successful in drawing links between their results and the importance biologically. The introduction does not clearly position this work in the context of previous literature, using relatively specialised technical terms without defining them, and leaving the reader unclear about how the results have advanced the field. In terms of their results, the authors further propose interesting links between LCRs and function. However, their analyses for these most exciting results rely heavily on UMAP visualisation and the use of tests with apparently small effect sizes. This is a weakness throughout the paper and reduces the support for strong conclusions.
Additionally, whilst the experimental work is interesting and concerns LCRs, it does not clearly fit into the rest of the body of work focused as it is on a single protein and the importance of its LCRs. It arguably serves as a validation of the method, but if that is the author's intention it needs to be made more clearly as it appears orthogonal to the overall drive of the paper.
Overall I think the ideas presented in the work are interesting, the method is sound, but the data does not clearly support the drawing of strong conclusions. The weakness in the conclusions and the poor description of the wider background lead me to question the impact of this work on the broader field.
Technical weaknesses
In the testing of the dotplot based method, the manuscript presents a FDR rate based on a comparison between real proteome data and a null proteome. This is a sensible approach, but their choice of a uniform random distribution would be expected to mislead. This is because if the distribution is non-uniform, stretches of the most frequent amino will occur more frequently than in the uniform distribution.
More generally I think the results presented suggest that the results dotplot generates are comparable to existing methods, not better and the text would be more accurate if this conclusion was clearer, in the absence of an additional set of data that could be used as a "ground truth".
The authors draw links between protein localisation/function and LCR content. This is done through the use of UMAP visualisation and wilcoxon rank sum tests on the amino acid frequency in different localisations. This is convincing in the case of ECM data, but the arguments are substantially less clear for other localisations/functions. The UMAP graphics show generally that the specific functions are sparsely spread. Moreover when considering the sample size (in the context of the whole proteome) the p-value threshold obscures what appear to be relatively small effect sizes.
-
Reviewer #2 (Public Review):
The authors present a novel dotplot-based approach to identify Low-complexity regions (LCRs) and dimensionality reduction techniques coupled with image processing methods to define the presence and organization of LCR regions. The dot-plot based method is unique in that it not only identifies different LCRs in a protein but could also define the relatedness between the different LCRs. Using UMAPs, they present a unified view of LCR sequence space in humans and predict the LCR space for different higher order assemblies. Using experimental studies they show how valency of LCRs could influence phase-separation behavior of such proteins. An expanded LCR sequence space map is used to capture the conservation and divergence of higher order assemblies LCR sequences across species. Finally, the authors showcase the …
Reviewer #2 (Public Review):
The authors present a novel dotplot-based approach to identify Low-complexity regions (LCRs) and dimensionality reduction techniques coupled with image processing methods to define the presence and organization of LCR regions. The dot-plot based method is unique in that it not only identifies different LCRs in a protein but could also define the relatedness between the different LCRs. Using UMAPs, they present a unified view of LCR sequence space in humans and predict the LCR space for different higher order assemblies. Using experimental studies they show how valency of LCRs could influence phase-separation behavior of such proteins. An expanded LCR sequence space map is used to capture the conservation and divergence of higher order assemblies LCR sequences across species. Finally, the authors showcase the efficacy of the approach by identifying a thus far unannotated T/H rich LCR cluster in Teleosts.
The method is robust. However, clarity is required regarding the FDR thresholding for identifying LCRs and assigning preponderant amino acids in the unified LCR sequence space. The findings regarding the sequence space across different higher order assemblies is commendable.
The approach and insights presented here could help all researchers working towards establishing protein sequence-function relationships and how variation in LCRs could affect fitness.
-
Reviewer #3 (Public Review):
The authors present a systematic assessment of low complexity sequences (LCRs) apply the dotplot matrix method for sequence comparison to identify low-complexity regions based on per-residue similarity. By taking the resulting self-comparison matrices and leveraging tools from image processing, the authors define LCRs based on similarity or non-similarity to one another. Taking the composition of these LCRs, the authors then compare how distinct regions of LCR sequence space compare across different proteomes.
The paper is well-written and easy to follow, and the results are consistent with prior work. The figures and data are presented in an extremely accessible way and the conclusions seem logical and sound.
My big picture concern stems from one that is perhaps challenging to evaluate, but it is not really …
Reviewer #3 (Public Review):
The authors present a systematic assessment of low complexity sequences (LCRs) apply the dotplot matrix method for sequence comparison to identify low-complexity regions based on per-residue similarity. By taking the resulting self-comparison matrices and leveraging tools from image processing, the authors define LCRs based on similarity or non-similarity to one another. Taking the composition of these LCRs, the authors then compare how distinct regions of LCR sequence space compare across different proteomes.
The paper is well-written and easy to follow, and the results are consistent with prior work. The figures and data are presented in an extremely accessible way and the conclusions seem logical and sound.
My big picture concern stems from one that is perhaps challenging to evaluate, but it is not really clear to me exactly what we learn here. The authors do a fine job of cataloging LCRs, offer a number of anecdotal inferences and observations are made - perhaps this is sufficient in terms of novelty and interest, but if anyone takes a proteome and identifies sequences based on some set of features that sit in the tails of the feature distribution, they can similarly construct intriguing but somewhat speculative hypotheses regarding the possible origins or meaning of those features.
The authors use the lysine-repeats as specific examples where they test a hypothesis, which is good, but the importance of lysine repeats in driving nucleolar localization is well established at this point - i.e. to me at least the bioinformatics analysis that precedes those results is unnecessary to have made the resulting prediction. Similarly, the authors find compositional biases in LCR proteins that are found in certain organelles, but those biases are also already established. These are not strictly criticisms, in that it's good that established patterns are found with this method, but I suppose my concern is that this is a lot of work that perhaps does not really push the needle particularly far.
As an important caveat to this somewhat muted reception, I recognize that having worked on problems in this area for 10+ years I may also be displaying my own biases, and perhaps things that are "already established" warrant repeating with a new approach and a new light. As such, this particular criticism may well be one that can and should be ignored.
That overall concern notwithstanding, I had several other questions that sprung to mind.
Dotplot matrix approach
The authors do a fantastic job of explaining this, but I'm left wondering, if one used an algorithm like (say) SEG, defined LCRs, and then compared between LCRs based on composition, would we expect the results to be so different? i.e. the authors make a big deal about the dotplot matrix approach enabling comparison of LCR type, but, it's not clear to me that this is just because it combines a two-step operation into a one-step operation. It would be useful I think to perform a similar analysis as is done later on using SEG and ask if the same UMAP structure appears (and discuss if yes/no).LCRs from repeat expansions
I did not see any discussion on the role that repeat expansions can play in defining LCRs. This seems like an important area that should be considered, especially if we expect certain LCRs to appear more frequently due to a combination of slippy codons and minimal impact due to the biochemical properties of the resulting LCR. The authors pursue a (very reasonable) model in which LCRs are functional and important, but it seems the alternative (that LCRs are simply an unavoidable product of large proteomes and emerge through genetic events that are insufficiently deleterious to be selected against). Some discussion on this would be helpful. it also makes me wonder if the authors' null proteome model is the "right" model, although I would also say developing an accurate and reasonable null model that accounts for repeat expansions is beyond what I would consider the scope of this paper.Minor points
Early on the authors discuss the roles of LCRs in higher-order assemblies. They then make reference to the lysine tracts as having a valence of 2 or 3. It is possibly useful to mention that valence reflects the number of simultaneous partners that a protein can interact with - while it is certainly possible that a single lysine tracts interacts with a single partner simultaneously (meaning the tract contributes a valence of 1) I don't think the authors can know that, so it may be wise to avoid specifying the specific valence.The authors make reference to Q/H LCRs. Recent work from Gutiérrez et al. eLife (2022) has argued that histidine-richness in some glutamine-rich LCRs is above the number expected based on codon bias, and may reflect a mode of pH sensing. This may be worth discussing.
Eric Ross has a number of very nice papers on this topic, but sadly I don't think any of them are cited here. On the question of LCR composition and condensate recruitment, I would recommend Boncella et al. PNAS (2020). On the question of proteome-wide LCR analysis, see Cascarina et al PLoS CompBio (2018) and Cascarina et al PLoS CompBio 2020.
-
