GWAS and functional studies suggest a role for altered DNA repair in the evolution of drug resistance in Mycobacterium tuberculosis
Curation statements for this article:-
Curated by eLife

eLife assessment
Drug-resistant tuberculosis (TB) is a growing threat to global public health. By analysing a large database of clinical Mycobacterium tuberculosis isolates, the authors of this study identify previously unrecognized genetic mutations that might be implicated in improved mycobacterial survival under antibiotic treatment. Using laboratory and experimental infection models, they present evidence that these mutations should be considered potential genetic markers of reduced antibiotic efficacy and accelerated acquisition of TB drug resistance.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
The emergence of drug resistance in Mycobacterium tuberculosis ( Mtb ) is alarming and demands in-depth knowledge for timely diagnosis. We performed genome-wide association analysis using 2237 clinical strains of Mtb to identify novel genetic factors that evoke drug resistance. In addition to the known direct targets, we identified for the first time, a strong association between mutations in DNA repair genes and the multidrug-resistant phenotype. To evaluate the impact of variants identified in the clinical samples in the evolution of drug resistance, we utilized knockouts and complemented strains in Mycobacterium smegmatis and Mtb . Results show that variant mutations compromised the functions of MutY and UvrB. MutY variant showed enhanced survival compared with wild-type ( Rv ) when the Mtb strains were subjected to multiple rounds of ex vivo antibiotic stress. In an in vivo guinea pig infection model, the MutY variant outcompeted the wild-type strain. We show that novel variant mutations in the DNA repair genes collectively compromise their functions and contribute to better survival under antibiotic/host stress conditions.
Article activity feed
-

-

Author Response:
Reviewer #1 (Public Review):
There is growing precedent for the utility of GWAS-type analyses in elucidating otherwise cryptic genotypic associations with specific Mtb phenotypes, most commonly drug resistance. This study represents the latest instalment of this type of approach, utilizing a large set of WGS data from clinical Mtb isolates and refining the search for DR-associated alleles by restricting the set to those predicted (or known) to be phenotypically DR. This revealed a number of potential candidate mutations, including some in nucleotide excision repair (uvrA, uvrB), in base excision repair (mutY), and homologous recombination (recF). In validating these leads functional assays, the authors present evidence supporting the impact of the identified mutations on antibiotic susceptibility in vitro and in …
Author Response:
Reviewer #1 (Public Review):
There is growing precedent for the utility of GWAS-type analyses in elucidating otherwise cryptic genotypic associations with specific Mtb phenotypes, most commonly drug resistance. This study represents the latest instalment of this type of approach, utilizing a large set of WGS data from clinical Mtb isolates and refining the search for DR-associated alleles by restricting the set to those predicted (or known) to be phenotypically DR. This revealed a number of potential candidate mutations, including some in nucleotide excision repair (uvrA, uvrB), in base excision repair (mutY), and homologous recombination (recF). In validating these leads functional assays, the authors present evidence supporting the impact of the identified mutations on antibiotic susceptibility in vitro and in macrophage and animal infection models. These results extend the number of candidate mutations associated with Mtb drug resistance, however the following must be considered:
(i) The GWAS analysis is the basis of this study, yet the description of the approach used and presentation of results obtained is occasionally obscure; for example, the authors report the use of known drug resistance phenotypes (where available) or inferences of drug-resistance from genotypic data to enhance the potential to identify other mutations that might be implicated in enabling the DR mutations, yet their list of known DR mutations seem to be predominantly rare or unusual mutations, not those commonly associated with clinical DR-TB. In addition, the distribution of the identified resistance-associated mutations across the different lineages need to be explained more clearly.
In the revised manuscript, we have performed the phylogenetic analysis of the strains used. A phylogenetic tree was generated using Mycobacterium canetti as an outgroup (Figure 1b). The phylogeny analysis suggests the clustering of the strains in lineage 1, 2, 3, and 4. Lineages 2,3 and 4 are clustering together, and lineage 1 is monophyletic, as reported previously. The genome sequence data of 2773 clinical strains were downloaded from NCBI. These strains were also part of the GWAS analysis performed by Coll et al (https://pubmed.ncbi.nlm.nih.gov/29358649/) and Manson et al. (https://pubmed.ncbi.nlm.nih.gov/28092681/). The phenotype of the strains used for the association analysis was reported in the previous studies. We have not performed other predictions. The supplementary table provides the lineage origin of each strain used in the study (Supplementary File 1 & 2). The distributions of resistance-associated mutations in different strains is shown (Figure 2-figure supplement 6a-h). As suggested, we have performed an analysis wherein we looked for the direct target mutations that harbor mutations in the DNA repair genes (Figure 2-figure supplement 6i-k).
We identified mostly the rare mutations due to the following reasons;
We looked for the mutations that were present only in the multidrug resistant strains as compared to the susceptible strains for association mapping. This strategy exclusively gave most variants associated with multidrug resistant phenotype.
We have used Mixed Linear Model (MLM) for association analysis. MLM removes all the population-specific SNPs based on PCA and kinship corrections. The false discovery rate (FDR) adjusted p-values in the GAPIT software are stringent as it corrects the effects of each marker based on the population structure (Q) as well as kinship (K) values. Therefore the probability of identifying the false-positive SNP is very low. We combined it with the Bonferroni corrections to identify markers associated with the drug resistant phenotype.
(ii) By combining target gene deletions with different complementation alleles, the authors provide compelling microbiological evidence supporting the inferred role of the mutY and uvrB mutations in enhanced survival under antibiotic treatment. The experimental work, however, is limited to assessments of competitive survival in various models, with/without antibiotic selection, or to mutant frequency analyses; there is no direct evidence provided in support of the proposed mechanism.
To ascertain if the better survival of the RvDmutY, or RvDmutY::mutY-R262Q, is indeed due to the acquisition of mutations in the direct target of antibiotics, we performed WGS of the strain from the ex vivo evolution experiment (Figure 5). Genomic DNA extracted from ten independent colonies (grown in vitro), was mixed in equal proportions before library preparation. Only those SNPs present in >20% of reads were retained for the analysis. Analysis of Rv sequences grown in vitro suggested that the laboratory strain has accumulated 100 SNPs compared with the reference strain. The sequence of Rv laboratory strain was used as the reference strain for the subsequent analysis. WGS data for RvDmutY, RvDmutY::mutY, and RvDmutY::mutY-R262Q strains grown in vitro did not show the presence of a mutation in the antibiotic target genes. In a similar vein, ten independent colonies, each from the 7H11-OADC plates, after the final round of ex vivo selection in the presence or absence of antibiotics, were selected for WGS. Data indicated that in the absence of antibiotics, no direct target mutations were identified in the ex vivo passaged strains (Figure 6a & e). In the presence of isoniazid, we found mutations in the katG (Ser315Thr or Ser315Ileu) in the Rv, RvDmutY but not in RvDmutY:mutY and RvDmutY::mutY-R262Q (Figure 6b & e). These findings are in congruence with the ex vivo evolution CFU analysis, wherein we did not observe a significant increase in the survival of RvDmutY and RvDmutY::mutY R262Q in the presence of isoniazid (Figure 5). In the presence of ciprofloxacin and rifampicin, direct target mutations were identified in the gyrA and rpoB (Figure 6c e). Asp94Glu/Asp94Gly mutations were identified in gyrA, and, His445Tyr/Ser450Leu mutations were identified in rpoB of RvDmutY and RvDmutY::mutY-R262Q, respectively. No direct target mutations were identified in the Rv and RvDmutY::mutY, suggesting that the perturbed DNA repair aids in acquiring the drug resistance-conferring mutations in Mtb (Figure 6c-e & Supplementary File 8).
To determine if the better survival of the RvDmutY, or RvDmutY::mutY-R262Q, in the guinea pig infection experiment (Figure 8) is due to the accumulation of mutations in the host, we performed WGS of the strain isolated from guinea pig lungs. Analysis revealed specific genes such as cobQ1, smc, espI, and valS were mutated only in RvDmutY and RvDmutY::mutYR262Q but not in Rv and RvDmutY::mutY. Besides, tcrA and gatA were mutated only in RvDmutY, whereas rv0746 were mutated exclusively in the RvDmutY:mutY (Figure 8-Figure Supplement 2). However, we did not observe any direct target mutations; this may be because guinea pigs were not subjected to antibiotic treatment. Data suggests that the continued longterm selection pressure is necessary for bacilli to acquire mutations.
(iii) The low drug concentrations used (especially of rifampicin against M. smegmatis) suggest the identified mutations confer low-level resistance to multiple antimycobacterial agents - in turn implying tolerance rather than resistance. If correct, it would be interesting to know how broadly tolerant strains containing these mutations are; that is, whether susceptibility is decreased to a broad range of antibiotics with different mechanisms of action (including both cidal and static agents), and whether the extent of the decrease be determined quantitatively (for example, as change in MIC value).
To evaluate the effect of different drugs on the survival of RvDmutY or RvDmutY::mutYR262Q, we performed killing kinetics in the presence and absence of isoniazid, rifampicin, ciprofloxacin, and ethambutol (Figure 4a). In the absence of antibiotics, the growth kinetics of Rv, RvDmutY, RvDmutY:mutY, and RvDmutY::mutY-R262Q were similar (Figure 4b). In the presence of isoniazid, ~2 log-fold decreases in bacterial survival was observed on day 3 in Rv and RvDmutY:mutY; however, in RvDmutY and RvDmutY::mutY-R262Q, the difference was limited to ~1.5 log-fold (Figure 4c). A similar trend was apparent on days 6 and 9, suggesting a ~5-fold increase in the survival of RvDmutY and RvDmutY::mutY-R262Q compared with Rv and RvDmutY:mutY (Figure 4c). Interestingly, in the presence of ethambutol, we did not observe any significant difference (Figure 4d). In the presence of rifampicin and ciprofloxacin, we observed a ~10-fold increase in the survival of RvDmutY and RvDmutY::mutY-R262Q compared with Rv and RvDmutY:mutY (Figure 4e-f). Thus results suggest that the absence of mutY or the presence of mutY variant aids in subverting the antibiotic stress.
Reviewer #2 (Public Review):
This interesting manuscript uses a collection of whole genome sequences of TB isolates to associate specific sequence polymorphisms with MDR/XDR strains, and having found certain mutations in DNA repair pathways, does a detailed analysis of several mutations. The evaluation of the MutY polymorphism reveals it is loss of function and TB strains carrying this mutation have a higher mutation frequency and enhanced survival in serial passage in macrophages. The strengths of the manuscript are the leveraging of a large sequence dataset to derive interesting candidate mutations in DNA repair pathway and the demonstration that at least one of these mutations has a detectable effect on mutagenicity and pathogenesis. The weaknesses of the manuscript are a lack of experimental exploration of the mechanism by which loss of a DNA repair pathway would enhance survival in vivo. The model presented is that these phenotypes are due to hypermutagenicity and thereby evolution of enhanced pathogenesis, but this is not actually directly tested or investigated. There are also some technical concerns for some of the experimental data which can be strengthened.
This paper presents the following data:
- Analyzed whole-genome sequences 2773 clinical strains: 160 000 SNPs identified
- 1815 drug-susceptible/422 MDR/XDR strains: 188 mutations correlated with Drug resistance.
- Novel mutations associated with the drug resistance have been found in base excision repair (BER), nucleotide excision repair (NER), and homologous recombination (HR) pathway genes (mutY, uvrA, uvrB, and recF).
- Specific mutations mutY-R262Q and uvrB-A524V were studied.
- mutY-R262Q and uvrB-A524V mutations behave as loss of function alleles in vivo, as measured by non-complementation of the increased mutation frequency measured by resistance to Rif and INH.
- The mutY deletion and the mutY-R262Q mutation increase Mtb survival over WT in macrophages when Mtb has not been submitted to previous rounds of macrophage infection.
- This advantage is exacerbated in presence of antibiotic (Rif and Cipro but not INH).
- The MutY deletion and the MutY-R262Q mutation result in an enhanced survival of Mtb during guinea pig infection.
Major issues:
The finding that mutations in MutY confers an advantage during macrophage infection is convincing based on the macrophage experiments, but it is premature to conclude that the mechanism of this effect is due to hypermutagenesis and selection of fitter bacterial clones. It is described in E. coli (Foti et al., 2012) and recently in mycobacteria (Dupuy et al., 2020) that the MutY/MutM excision pathways can increase the lethality of antibiotic treatment because of double-strand breaks caused by Adenine/oxoG excisions. The higher survival of the mutY mutant during antibiotic treatment could more be due to lower Adenine/oxoG excision in the mutant rather than acquisition of advantageous mutations, or some other mechanism. The same hypothesis cannot be excluded for the Guinea pig experiments (no antibiotics, but oxidative stress mediated by host defenses could also increase oxoG) and should at least be discussed. Experiments that would support the idea that the in vivo advantage is due to hypermutagenesis would be whole genome sequencing of the output vs input populations to directly document increased mutagenesis. Similarly, is the ΔmutY survival advantage after rounds of macrophage infections dependent on macrophage environment? What happens if the ΔmutY strain is cultivated in vitro in 7H9 (same number of generations) before infecting macrophages?
We thank the reviewer for the insightful comments. To ascertain if the better survival of the RvDmutY, or RvDmutY::mutY-R262Q, is indeed due to the acquisition of mutations in the direct target of antibiotics, we performed WGS of the strain from the ex vivo evolution experiment (Figure 5). Genomic DNA extracted from ten independent colonies (grown in vitro) was mixed in equal proportion prior to library preparation. For the analysis, only those SNPs that were present in >20% of reads were retained. Analysis of Rv sequences grown in vitro suggested that the laboratory strain has accumulated 100 SNPs compared with the reference strain. The sequence of the Rv laboratory strain was used as the reference strain for the subsequent analysis. WGS data for RvDmutY, RvDmutY::mutY, and RvDmutY::mutY-R262Q strains grown in vitro did not show the presence of a mutation in the antibiotic target genes. In a similar vein, ten independent colonies, each from the 7H11-OADC plates, after the final round of ex vivo selection in the presence or absence of antibiotics, were selected for WGS. Data indicated that in the absence of antibiotic, no direct target mutations were identified in the ex vivo passaged strains (Figure 6a & e). In the presence of isoniazid, we found mutations in the katG (Ser315Thr or Ser315Ileu) in the Rv, RvDmutY but not in RvDmutY:mutY and RvDmutY::mutY-R262Q (Figure 6b & e). These findings are in congruence with the ex vivo evolution CFU analysis, wherein we did not observe a significant increase in the survival of RvDmutY and RvDmutY::mutY R262Q in the presence of isoniazid (Figure 5). In the presence of ciprofloxacin and rifampicin, direct target mutations were identified in the gyrA and rpoB (Figure 6c-e). Asp94Glu/Asp94Gly mutations were identified in gyrA, and, His445Tyr/Ser450Leu mutations were identified in rpoB of RvDmutY and RvDmutY::mutY-R262Q, respectively. No direct target mutations were identified in the Rv and RvDmutY::mutY, suggesting that the perturbed DNA repair aids in acquiring the drug resistance-conferring mutations in Mtb (Figure 6c-e & Supplementary File 8).
To determine if the better survival of the RvDmutY, or RvDmutY::mutY-R262Q, in the guinea pig infection experiment (Figure 8) is due to the accumulation of mutations in the host, we performed WGS of the strain isolated from guinea pig lungs. Analysis revealed specific genes such as cobQ1, smc, espI, and valS were mutated only in RvDmutY and RvDmutY::mutYR262Q but not in Rv and RvDmutY::mutY. Besides, tcrA and gatA were mutated only in RvDmutY, whereas rv0746 were mutated exclusively in the RvDmutY:mutY (Figure 8-figure supplement 2). However, we did not observe any direct target mutations; this may be because guinea pigs were not subjected to antibiotic treatment. Data suggests that the continued longterm selection pressure is necessary for bacilli to acquire mutations.
- It would be useful to present more data about the strain relatedness and genome characteristics of the DNA repair mutant strains in the GWAS. For example, the model would suggest that strains carrying DNA repair mutations should have higher SNP load than control strains. Additionally, it would be helpful to know whether the identified DNA repair pathway mutations are from epidemiologically linked strains in the collection to deduce whether these events are arising repeatedly or are a founder effect of a single mutant since for each mutation, the number of strains is small.
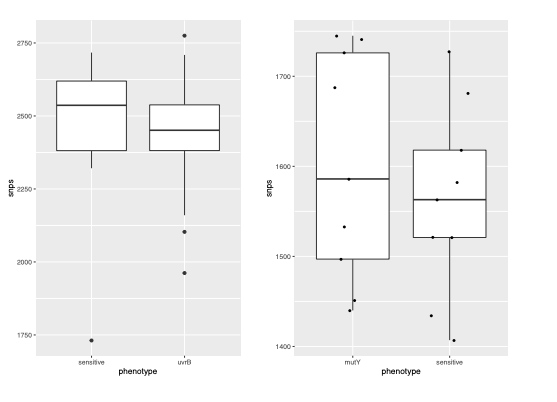
We analyzed the genome of the clinical strains that possess DNA repair gene mutations to determine the additional polymorphisms. The number of SNPs in the strains harboring DNA repair mutation and the drug susceptible strains appears to be similar. The marginal difference, if any were not statistically significant.
We agree with the reviewer that these strains might be epidemiologically linked. In the present study, all the strains harboring mutation in mutY belong to lineage 4. We observed that all the mutY mutationcontaining strains were either MDR or pre-XDR compared with drug susceptible strains of the same clade.
- Some of the mutation frequency, survival and competition data could be strengthened by more experimental replicates. Data Lines 370-372 (mutation frequency), lines 387-388 (Survival of strains ex vivo), line 394 (competition experiment) : "Two biologically independent experiments were performed. Each experiment was performed in technical triplicates. Data represent one of the two biological experiments." Two biological replicates is insufficient for the phenotypes presented and all replicates should be included in the analysis. In addition, the definition of "technical triplicates" should be given, does this mean the same culture sampled in triplicate?
We thank the reviewer for the comment. We performed at least two independent experiments with biological triplicates (not technical triplicates). We apologize for writing this incorrectly. We have reported data from one independent experiment consisting of at least biological triplicates. For mutation rate analysis, we have performed experiment using six independent colonies. These points are mentioned in the methods and legends of the revised manuscript.
- MutY phenotypes. One caveat to the conclusion that the MutY R262Q mutant is nonfunctional is the lack of examination of the expression of the complementing protein. I would be informative to comment on the location of this mutation in relation to the known structures of MutY proteins. Similarly, for the UvrB polymorphism, this null strain has a clear UV sensitivity phenotype in the literature, so a fuller interrogation for UV killing would be informative re: the A524V mutation.
We have now included the western blot data on both complementation strains (Figure 3-figure supplement 1). We agree with the reviewer that the uvrB null mutant may have UV sensitivity phenotype, but we have not performed the experiment in the present study.
Reviewer #3 (Public Review):
STRENGTHS
• This ambitious study is broad in scope, beginning with a bacterial GWAS study and extending all the way to in vivo guinea pig infection models.
• Numerous reports have attempted to identify Mtb strains with elevated mutation rates, and the results are conflicting. The present study sets out to thoroughly evaluate one such mutation that may produce a mutator phenotype, mutY-Arg262Gln.
WEAKNESSES
• While the authors follow-up experiments with the mutY-Arg262Gln allele are all consistent with the conclusion that this mutation elevates the mutation rate in Mtb and thus could promote the evolution of drug resistance, further work is needed to unambiguously demonstrate this link.
• The authors highlight five mutations in genes associated with DNA replication and or repair from their GWAS analysis:
o dnaA-Arg233Gln: as the authors note in the Discussion, Hicks et al. associate SNPs in dnaA with low-level isoniazid resistance, as a result of lowered katG expression. Since this is unrelated to their focus on DNA repair genes whose mutation could elevate mutation rates, I would consider removing this allele from the Table.
As suggested, we have removed the dnaA from Table 3.
o mutY-Arg262Gln: querying publicly available whole genome sequences of clinical Mtb isolates, this SNP appears to be restricted to lineage 4.3 (L4.3). All of these L4.3 strains appear to be drug-resistant. How many times did the mutY-Arg262Gln mutation evolve in the authors dataset? If there is evidence of homoplastic evolution, this would strengthen their case. If not, it doesn't mean the authors findings are incorrect, but does elevate that risk that this mutation could be a passenger (i.e. not driver) mutation. To address this, the authors could attempt to date when the mutY-Arg262Gln arose. If it was before the evolution of drug-resistance conferring alleles in these L4.3 strains, that is consistent with (but not proof of) a driver mutation. If mutY-Arg262Gln arose after, this is much more consistent with a passenger mutation.
As pointed out by the reviewer, the mutY-Arg262Gln mutation is restricted to lineage 4. We have checked the mutY gene sequence from the strains harboring mutY Arg262Gln mutation and sensitive strains of the same clade. We identified only the reported mutation in the drug-resistant strains, and there was no synonymous mutation that could be used for performing molecular clock analysis. To ascertain whether it is a passenger or a driver mutation, we have performed multiple experiments that suggest that identified mutation aids in the acquisition of drug resistance.
o uvrB-Ala524Val: curiously we don't see this SNP in our dataset of publicly available whole genome sequences of clinical Mtb isolates (~45,000 genomes).
We have rechecked this SNP in our dataset. This SNP was present in 87 drug-resistant strains that belong to lineage 2.
o uvrA-Gln135Lys: this SNP also appears to be restricted to lineage 4.3. Same question as for mutY-Arg262Gln.
As pointed out by the reviewer, uvrA-Gln135lys mutation is restricted to lineage 4. We identified only the reported mutation in the drug-resistant strains, and there was no synonymous mutation that can be used for performing molecular clock analysis
o recF-Gly269Gly: this is a very common mutation, is it unique to lineage 2.2.1? Same question as for mutY-Arg262Gln.
RecF-Gly269Gly mutation was present in the lineage 2 strains. Here also, we identified only the reported mutation in the drug-resistant strains, and there was no synonymous mutation could be used for performing molecular clock analysis.
• The CRYPTIC consortium recently published a number of preprints on biorxiv detailing very large GWAS studies in Mtb. Did any of these reports also associate drug resistance with mutY? If yes, this should be stated. If not, the potential reasons for this discrepancy should be discussed.
We have checked the recently published CRYPTIC consortium article (https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3001721#sec012) for mutY-Arg262Gln. We did not find the mutY-Arg262Gln mutation in their analysis; this is due to the different strains used in the study. However, we identified recF Gly269Gly mutation in their datase
• Based on the authors follow-up studies in vivo, MutY-Arg262Gln is presumed to be a loss-of-function allele. If the authors could convincingly demonstrate this biochemically with recombinant proteins, this would significantly strengthen their case.
Experiments performed in Msm and Mtb mutant strains suggest that MutY variant is a loss-of-function allele. We have not performed in vitro assays to confirm the same.
• If the authors are correct and mutY-Arg262Gln strains have elevated mutation rates, presumably there would be evidence of this in the clinical strain sequencing data. Do mutY-Arg262Gln containing strains have elevated C→G or C→A mutations in their genomes? Presumably such strains would also have a higher number of SNPs than closely related strains WT for mutY- is this the case?
We analyzed the genome of the clinical strains that possess DNA repair gene mutations to determine the additional polymorphisms. The number of SNPs in the strains harboring DNA repair mutation and the drug susceptible strains appears to be higher. We have also looked for the CàT and CàG mutations in the same strains. CàT mutations are higher in the strains harboring mutY variant compared with the susceptible strains (Figure 2-figure supplement 6 l). However, we could not perform statistical analysis as the number of strains that harbor mutY variant is limited to 8. Thus data suggest that empirically the strains harboring mutY variant show higher SNPs elsewhere and CàT mutations. We are not stating these conclusions strongly in the manuscript as the data is not statistically significant
• While more work, mutation rates as measured by Luria-Delbruck fluctuation analysis are more accurate than mutation frequencies. I would recommend repeating key experiments by Luria-Delbruck fluctuation analysis. It is also important to report both drug-resistant colony counts and total CFU in these sorts of experiments. Given the clumpy nature of mycobacteria, mutation rates can appear to be artificially elevated due to low total CFU and not an increase in the number of drug-resistant colonies.
As suggested, we determined the mutation rate in the presence of isoniazid, rifampicin, and ciprofloxacin (Figure 3g-j). The fold increase in the mutation rate relative to Rv for RvDmutY, RvDmutY:mutY, and RvDmutY::mutY-R262Q was 2.90, 0.76, and 3.0 in the presence of isoniazid and 5.62, 1.13, and 5.10 or 9.14, 1.57, and 8.71 in the presence of rifampicin and ciprofloxacin respectively (Figure 3).
• Figure 4 would appear to measuring drug tolerance not resistance? Are the elevated CFU in the presence of drugs in the mutY-Arg262Gln strain due to an increase in the number of drug resistant strains or drug sensitive strains? This could be assessed by quantifying resulting CFU in the presence or absence the indicated drugs.
To ascertain better survival is due to the acquisition of mutations in the direct target of antibiotics or drug tolerance. We performed WGS of the strain from the ex vivo evolution experiment (Figure 5). Genomic DNA extracted from ten independent colonies (grown in vitro) was mixed in equal proportion prior to library preparation. Only those SNPs present in >20% of reads were retained for the analysis. Analysis of Rv sequences grown in vitro suggested that the laboratory strain has accumulated 100 SNPs compared with the reference strain. The sequence of the Rv laboratory strain was used as the reference strain for the subsequent analysis. WGS data for RvDmutY, RvDmutY::mutY, and RvDmutY::mutY-R262Q strains grown in vitro did not show the presence of a mutation in the antibiotic target genes. In a similar vein, ten independent colonies, each from the 7H11-OADC plates, after the final round of ex vivo selection in the presence or absence of antibiotics, were selected for WGS. Data indicated that in the absence of antibiotics, no direct target mutations were identified in the ex vivo passaged strains (Figure 6a & e). In the presence of isoniazid, we found mutations in the katG (Ser315Thr or Ser315Ileu) in the Rv, RvDmutY but not in RvDmutY::mutY and RvDmutY::mutY-R262Q (Figure 6b & e). These findings are in congruence with the ex vivo evolution CFU analysis, wherein we did not observe a significant increase in the survival of RvDmutY and RvDmutY::mutY-R262Q in the presence of isoniazid (Figure 5). In the presence of ciprofloxacin and rifampicin, direct target mutations were identified in the gyrA and rpoB (Figure 6c-e). Asp94Glu/Asp94Gly mutations were identified in gyrA, and, His445Tyr/Ser450Leu mutations were identified in rpoB of RvDmutY and RvDmutY::mutY-R262Q, respectively. No direct target mutations were identified in the Rv and RvDmutY::mutY, suggesting that the perturbed DNA repair aids in acquiring the drug resistance-conferring mutations in Mtb (Figure 6c-e & Supplementary File 8).
To determine if the better survival of the RvDmutY, or RvDmutY::mutY-R262Q, in the guinea pig infection experiment (Figure 8) is due to the accumulation of mutations in the host, we performed WGS of the strain isolated from guinea pig lungs. Analysis revealed specific genes such as cobQ1, smc, espI, and valS were mutated only in RvDmutY and RvDmutY::mutYR262Q but not in Rv and RvDmutY::mutY. Besides, tcrA and gatA were mutated only in RvDmutY, whereas rv0746 were mutated exclusively in the RvDmutY::mutY (Figure 2-figure supplement 6). However, we did not observe any direct target mutations; this may be because guinea pigs were not subjected to antibiotic treatment. Data suggests that the continued longterm selection pressure is necessary for bacilli to acquire mutations.
-
eLife assessment
Drug-resistant tuberculosis (TB) is a growing threat to global public health. By analysing a large database of clinical Mycobacterium tuberculosis isolates, the authors of this study identify previously unrecognized genetic mutations that might be implicated in improved mycobacterial survival under antibiotic treatment. Using laboratory and experimental infection models, they present evidence that these mutations should be considered potential genetic markers of reduced antibiotic efficacy and accelerated acquisition of TB drug resistance.
-
Reviewer #1 (Public Review):
There is growing precedent for the utility of GWAS-type analyses in elucidating otherwise cryptic genotypic associations with specific Mtb phenotypes, most commonly drug resistance. This study represents the latest instalment of this type of approach, utilizing a large set of WGS data from clinical Mtb isolates and refining the search for DR-associated alleles by restricting the set to those predicted (or known) to be phenotypically DR. This revealed a number of potential candidate mutations, including some in nucleotide excision repair (uvrA, uvrB), in base excision repair (mutY), and homologous recombination (recF). In validating these leads functional assays, the authors present evidence supporting the impact of the identified mutations on antibiotic susceptibility in vitro and in macrophage and animal …
Reviewer #1 (Public Review):
There is growing precedent for the utility of GWAS-type analyses in elucidating otherwise cryptic genotypic associations with specific Mtb phenotypes, most commonly drug resistance. This study represents the latest instalment of this type of approach, utilizing a large set of WGS data from clinical Mtb isolates and refining the search for DR-associated alleles by restricting the set to those predicted (or known) to be phenotypically DR. This revealed a number of potential candidate mutations, including some in nucleotide excision repair (uvrA, uvrB), in base excision repair (mutY), and homologous recombination (recF). In validating these leads functional assays, the authors present evidence supporting the impact of the identified mutations on antibiotic susceptibility in vitro and in macrophage and animal infection models. These results extend the number of candidate mutations associated with Mtb drug resistance, however the following must be considered:
(i) The GWAS analysis is the basis of this study, yet the description of the approach used and presentation of results obtained is occasionally obscure; for example, the authors report the use of known drug resistance phenotypes (where available) or inferences of drug-resistance from genotypic data to enhance the potential to identify other mutations that might be implicated in enabling the DR mutations, yet their list of known DR mutations seem to be predominantly rare or unusual mutations, not those commonly associated with clinical DR-TB. In addition, the distribution of the identified resistance-associated mutations across the different lineages need to be explained more clearly.
(ii) By combining target gene deletions with different complementation alleles, the authors provide compelling microbiological evidence supporting the inferred role of the mutY and uvrB mutations in enhanced survival under antibiotic treatment. The experimental work, however, is limited to assessments of competitive survival in various models, with/without antibiotic selection, or to mutant frequency analyses; there is no direct evidence provided in support of the proposed mechanism.
(iii) The low drug concentrations used (especially of rifampicin against M. smegmatis) suggest the identified mutations confer low-level resistance to multiple antimycobacterial agents - in turn implying tolerance rather than resistance. If correct, it would be interesting to know how broadly tolerant strains containing these mutations are; that is, whether susceptibility is decreased to a broad range of antibiotics with different mechanisms of action (including both cidal and static agents), and whether the extent of the decrease be determined quantitatively (for example, as change in MIC value).
-
Reviewer #2 (Public Review):
This interesting manuscript uses a collection of whole genome sequences of TB isolates to associate specific sequence polymorphisms with MDR/XDR strains, and having found certain mutations in DNA repair pathways, does a detailed analysis of several mutations. The evaluation of the MutY polymorphism reveals it is loss of function and TB strains carrying this mutation have a higher mutation frequency and enhanced survival in serial passage in macrophages. The strengths of the manuscript are the leveraging of a large sequence dataset to derive interesting candidate mutations in DNA repair pathway and the demonstration that at least one of these mutations has a detectable effect on mutagenicity and pathogenesis. The weaknesses of the manuscript are a lack of experimental exploration of the mechanism by which …
Reviewer #2 (Public Review):
This interesting manuscript uses a collection of whole genome sequences of TB isolates to associate specific sequence polymorphisms with MDR/XDR strains, and having found certain mutations in DNA repair pathways, does a detailed analysis of several mutations. The evaluation of the MutY polymorphism reveals it is loss of function and TB strains carrying this mutation have a higher mutation frequency and enhanced survival in serial passage in macrophages. The strengths of the manuscript are the leveraging of a large sequence dataset to derive interesting candidate mutations in DNA repair pathway and the demonstration that at least one of these mutations has a detectable effect on mutagenicity and pathogenesis. The weaknesses of the manuscript are a lack of experimental exploration of the mechanism by which loss of a DNA repair pathway would enhance survival in vivo. The model presented is that these phenotypes are due to hypermutagenicity and thereby evolution of enhanced pathogenesis, but this is not actually directly tested or investigated. There are also some technical concerns for some of the experimental data which can be strengthened.
This paper presents the following data:
- Analyzed whole-genome sequences 2773 clinical strains: 160 000 SNPs identified
- 1815 drug-susceptible/422 MDR/XDR strains: 188 mutations correlated with Drug resistance.
- Novel mutations associated with the drug resistance have been found in base excision repair (BER), nucleotide excision repair (NER), and homologous recombination (HR) pathway genes (mutY, uvrA, uvrB, and recF).
- Specific mutations mutY-R262Q and uvrB-A524V were studied.
- mutY-R262Q and uvrB-A524V mutations behave as loss of function alleles in vivo, as measured by non-complementation of the increased mutation frequency measured by resistance to Rif and INH.
- The mutY deletion and the mutY-R262Q mutation increase Mtb survival over WT in macrophages when Mtb has not been submitted to previous rounds of macrophage infection.
- This advantage is exacerbated in presence of antibiotic (Rif and Cipro but not INH).
- The MutY deletion and the MutY-R262Q mutation result in an enhanced survival of Mtb during guinea pig infection.Major issues:
The finding that mutations in MutY confers an advantage during macrophage infection is convincing based on the macrophage experiments, but it is premature to conclude that the mechanism of this effect is due to hypermutagenesis and selection of fitter bacterial clones. It is described in E. coli (Foti et al., 2012) and recently in mycobacteria (Dupuy et al., 2020) that the MutY/MutM excision pathways can increase the lethality of antibiotic treatment because of double-strand breaks caused by Adenine/oxoG excisions. The higher survival of the mutY mutant during antibiotic treatment could more be due to lower Adenine/oxoG excision in the mutant rather than acquisition of advantageous mutations, or some other mechanism. The same hypothesis cannot be excluded for the Guinea pig experiments (no antibiotics, but oxidative stress mediated by host defenses could also increase oxoG) and should at least be discussed. Experiments that would support the idea that the in vivo advantage is due to hypermutagenesis would be whole genome sequencing of the output vs input populations to directly document increased mutagenesis. Similarly, is the ΔmutY survival advantage after rounds of macrophage infections dependent on macrophage environment? What happens if the ΔmutY strain is cultivated in vitro in 7H9 (same number of generations) before infecting macrophages?
- It would be useful to present more data about the strain relatedness and genome characteristics of the DNA repair mutant strains in the GWAS. For example, the model would suggest that strains carrying DNA repair mutations should have higher SNP load than control strains. Additionally, it would be helpful to know whether the identified DNA repair pathway mutations are from epidemiologically linked strains in the collection to deduce whether these events are arising repeatedly or are a founder effect of a single mutant since for each mutation, the number of strains is small.
- Some of the mutation frequency, survival and competition data could be strengthened by more experimental replicates. Data Lines 370-372 (mutation frequency), lines 387-388 (Survival of strains ex vivo), line 394 (competition experiment) : "Two biologically independent experiments were performed. Each experiment was performed in technical triplicates. Data represent one of the two biological experiments." Two biological replicates is insufficient for the phenotypes presented and all replicates should be included in the analysis. In addition, the definition of "technical triplicates" should be given, does this mean the same culture sampled in triplicate?
- MutY phenotypes. One caveat to the conclusion that the MutY R262Q mutant is nonfunctional is the lack of examination of the expression of the complementing protein. I would be informative to comment on the location of this mutation in relation to the known structures of MutY proteins. Similarly, for the UvrB polymorphism, this null strain has a clear UV sensitivity phenotype in the literature, so a fuller interrogation for UV killing would be informative re: the A524V mutation.
-
Reviewer #3 (Public Review):
STRENGTHS
• This ambitious study is broad in scope, beginning with a bacterial GWAS study and extending all the way to in vivo guinea pig infection models.
• Numerous reports have attempted to identify Mtb strains with elevated mutation rates, and the results are conflicting. The present study sets out to thoroughly evaluate one such mutation that may produce a mutator phenotype, mutY-Arg262Gln.
WEAKNESSES
• While the authors follow-up experiments with the mutY-Arg262Gln allele are all consistent with the conclusion that this mutation elevates the mutation rate in Mtb and thus could promote the evolution of drug resistance, further work is needed to unambiguously demonstrate this link.
• The authors highlight five mutations in genes associated with DNA replication and or repair from their GWAS analysis:
o …
Reviewer #3 (Public Review):
STRENGTHS
• This ambitious study is broad in scope, beginning with a bacterial GWAS study and extending all the way to in vivo guinea pig infection models.
• Numerous reports have attempted to identify Mtb strains with elevated mutation rates, and the results are conflicting. The present study sets out to thoroughly evaluate one such mutation that may produce a mutator phenotype, mutY-Arg262Gln.
WEAKNESSES
• While the authors follow-up experiments with the mutY-Arg262Gln allele are all consistent with the conclusion that this mutation elevates the mutation rate in Mtb and thus could promote the evolution of drug resistance, further work is needed to unambiguously demonstrate this link.
• The authors highlight five mutations in genes associated with DNA replication and or repair from their GWAS analysis:
o dnaA-Arg233Gln: as the authors note in the Discussion, Hicks et al. associate SNPs in dnaA with low-level isoniazid resistance, as a result of lowered katG expression. Since this is unrelated to their focus on DNA repair genes whose mutation could elevate mutation rates, I would consider removing this allele from the Table.
o mutY-Arg262Gln: querying publicly available whole genome sequences of clinical Mtb isolates, this SNP appears to be restricted to lineage 4.3 (L4.3). All of these L4.3 strains appear to be drug-resistant. How many times did the mutY-Arg262Gln mutation evolve in the authors dataset? If there is evidence of homoplastic evolution, this would strengthen their case. If not, it doesn't mean the authors findings are incorrect, but does elevate that risk that this mutation could be a passenger (i.e. not driver) mutation. To address this, the authors could attempt to date when the mutY-Arg262Gln arose. If it was before the evolution of drug-resistance conferring alleles in these L4.3 strains, that is consistent with (but not proof of) a driver mutation. If mutY-Arg262Gln arose after, this is much more consistent with a passenger mutation.
o uvrB-Ala524Val: curiously we don't see this SNP in our dataset of publicly available whole genome sequences of clinical Mtb isolates (~45,000 genomes).
o uvrA-Gln135Lys: this SNP also appears to be restricted to lineage 4.3. Same question as for mutY-Arg262Gln.
o recF-Gly269Gly: this is a very common mutation, is it unique to lineage 2.2.1? Same question as for mutY-Arg262Gln.
• The CRYPTIC consortium recently published a number of preprints on biorxiv detailing very large GWAS studies in Mtb. Did any of these reports also associate drug resistance with mutY? If yes, this should be stated. If not, the potential reasons for this discrepancy should be discussed.
• Based on the authors follow-up studies in vivo, MutY-Arg262Gln is presumed to be a loss-of-function allele. If the authors could convincingly demonstrate this biochemically with recombinant proteins, this would significantly strengthen their case.
• If the authors are correct and mutY-Arg262Gln strains have elevated mutation rates, presumably there would be evidence of this in the clinical strain sequencing data. Do mutY-Arg262Gln containing strains have elevated C→G or C→A mutations in their genomes? Presumably such strains would also have a higher number of SNPs than closely related strains WT for mutY- is this the case?
• While more work, mutation rates as measured by Luria-Delbruck fluctuation analysis are more accurate than mutation frequencies. I would recommend repeating key experiments by Luria-Delbruck fluctuation analysis. It is also important to report both drug-resistant colony counts and total CFU in these sorts of experiments. Given the clumpy nature of mycobacteria, mutation rates can appear to be artificially elevated due to low total CFU and not an increase in the number of drug-resistant colonies.
• Figure 4 would appear to measuring drug tolerance not resistance? Are the elevated CFU in the presence of drugs in the mutY-Arg262Gln strain due to an increase in the number of drug resistant strains or drug sensitive strains? This could be assessed by quantifying resulting CFU in the presence or absence the indicated drugs.
-
