Complex fitness landscape shapes variation in a hyperpolymorphic species
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
Stolyarova et al. investigate a highly polymorphic species, the fungus Schizophyllum commune, finding that compared to synonymous mutations, levels of linkage disequilibrium between nonsynonymous mutations are higher within genes than between genes. The authors propose this observation may be explained by compensatory interactions between nonsynonymous alleles, pointing to the presence of positive epistasis. This paper should be of interest to population geneticists and evolutionary biologists studying the role of natural selection.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. The reviewers remained anonymous to the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
It is natural to assume that patterns of genetic variation in hyperpolymorphic species can reveal large-scale properties of the fitness landscape that are hard to detect by studying species with ordinary levels of genetic variation. Here, we study such patterns in a fungus Schizophyllum commune , the most polymorphic species known. Throughout the genome, short-range linkage disequilibrium (LD) caused by attraction of minor alleles is higher between pairs of nonsynonymous than of synonymous variants. This effect is especially pronounced for pairs of sites that are located within the same gene, especially if a large fraction of the gene is covered by haploblocks, genome segments where the gene pool consists of two highly divergent haplotypes, which is a signature of balancing selection. Haploblocks are usually shorter than 1000 nucleotides, and collectively cover about 10% of the S. commune genome. LD tends to be substantially higher for pairs of nonsynonymous variants encoding amino acids that interact within the protein. There is a substantial correlation between LDs at the same pairs of nonsynonymous mutations in the USA and the Russian populations. These patterns indicate that selection in S. commune involves positive epistasis due to compensatory interactions between nonsynonymous alleles. When less polymorphic species are studied, analogous patterns can be detected only through interspecific comparisons.
Article activity feed
-

-

Author Response:
Reviewer #1 (Public Review):
Two important goals in evolutionary biology are (i) to understand why different species exhibit different levels of genetic diversity and (ii) in each species, what is the evolutionary nature of genetic variants. Are genetic variants mostly neutral, deleterious, or advantageous? In their study, Stolyarova et al. looked at one of the most polymorphic species known, the fungus Schizophyllum commune. They found that in this hyperpolymorphic species, the evolutionary forces that govern and structure genetic variation can be very different compared to less polymorphic species, including humans and flies. Specifically, the authors find that a process known as positive epistasis is quantitatively abundant among genetic variants that alter proteins in S. commune. Positive epistasis happens when a …
Author Response:
Reviewer #1 (Public Review):
Two important goals in evolutionary biology are (i) to understand why different species exhibit different levels of genetic diversity and (ii) in each species, what is the evolutionary nature of genetic variants. Are genetic variants mostly neutral, deleterious, or advantageous? In their study, Stolyarova et al. looked at one of the most polymorphic species known, the fungus Schizophyllum commune. They found that in this hyperpolymorphic species, the evolutionary forces that govern and structure genetic variation can be very different compared to less polymorphic species, including humans and flies. Specifically, the authors find that a process known as positive epistasis is quantitatively abundant among genetic variants that alter proteins in S. commune. Positive epistasis happens when a combination of multiple genetic variants is advantageous for the individuals that carry them, even though each isolated variant in the combination is not advantageous or even detrimental on its own. The authors explain that this happens frequently in their hyperpolymorphic species because the very high polymorphism level makes it very likely that the genetic variants will by chance occur together in the same individuals. In less polymorphic species, the variants that are advantageous in combination may have to wait for each other to occur for too long, for the combination to ever happen often enough in the first place.
Overall I had a great time reading the manuscript, and I feel that my understanding of evolution has been advanced on a fundamental level after reading it. However part of the reason why I enjoyed it was having to fill the gaps, answer the riddles left unanswered in the story by the authors.
Strengths:
- The model, both extremely polymorphic and amenable to haploid cultures, is ideal to address the questions asked.
- The study potentially represents a very important conceptual advance on the way to better understand genetic variation in general.
- The interpretations made by the authors of their data are likely the correct ones to make, even though more definitive answers will likely only come from the sequencing of a much larger number of haplotypes, which cannot reasonably be asked of the authors at this point.
Weaknesses:
- The manuscript does not provide enough information to judge if the synonymous controls that are compared to the nonsynonymous variants are fully adequate. Specifically, I have one concern that the Site Frequency Spectrum (SFS) of the synonymous variants at MAF>0.05 may be very different compared to the SFS of nonsynonymous variants at MAF>0.05. I focus on this because the authors mention page 5 line 3: "The excess of LDnonsyn over LDsyn corresponds to the attraction between rare alleles at nonsynonymous sites". First, it is unclear from this or from the figures at this point in the manuscript what the authors mean by rare alleles, among those alleles at MAF>0.05. This needs to be detailed quantitatively much more carefully. Second, and most importantly, this raises the question of whether or not the synonymous controls have a SFS with many less rare (but with MAF>0.05) alleles, as one may expect if they are under less purifying selection than nonsynonymous variants. This then raises the question of whether or not the synonymous control conducted by the author is adequate, or if the authors need to explicitly match the synonymous control in terms of SFS for MAF>0.05 in addition to the distance matching already done.
We thank the reviewer for this important comment. In page 5 line 3 we meant “the attraction between minor alleles”. In order to avoid confusion between SNPs with low MAF (“rare”) and minor variants at these polymorphic sites (“minor” ) we replaced “rare alleles” with “minor alleles” where appropriate.
The attraction between minor alleles in nonsynonymous polymorphic sites in S. commune holds if we pool all SNPs together, as is shown in Figure 2 - supplementary figure 4. Following the reviewer’s suggestion, we performed an additional analysis of LD between frequency-matched synonymous and nonsynonymous pairs of SNPs. Specifically, for each possible minor allele count and nucleotide distance, we calculated the number of corresponding pairs of nonsynonymous SNPs and subsampled the same number of synonymous SNPs with the same minor allele count and nucleotide distance. Such subsampling with exact matching of both MAFs and distance shows that LDnonsyn is elevated as compared to LDsyn in both S. commune populations (Figure 2 - figure supplement 3 of the revised version of the manuscript).
- The manuscript is far too succinct on several occasions, where observations or interpretations need to be much more detailed and explained.
We revised the manuscript for clarity, as detailied below.
Reviewer #2 (Public Review):
Stolyarova et al. used a highly polymorphic species, Schizophyllum commune, to explore patterns of LD between nonsynonymous and synonymous mutations within protein-coding genes. LD is informative about interference and interactions between selected loci, with compensatory mutations expected to be in strong positive LD. The benefit of studying this fungal species with large diversity (with pi > 0.1) is that populations are able to explore relatively large regions of the fitness landscape, and chances increase that sets of epistatically interacting mutations segregate at the same time.
This study finds strong positive LD between pairs of nonsynonymous mutations within, but not between genes, compared to pairs synonymous variants. Further, the authors show that high LD is prevalent among pairs of mutations at amino acid sites that interact within the protein. This result is consistent with pairs or sets of compensatory nonsynonymous mutations cosegregating within protein-coding genes.
The conclusions of this paper are largely supported by the data, with some caveats, listed below.
- With such large pairwise diversity, there are bound to be many deleterious variants segregating at once, and the large levels of interference between them will make selection much less efficient at purging deleterious variants.
We agree that simultaneous segregation of multiple deleterious nonsynonymous variants in the linked locus impedes their elimination by negative selection. However, stronger Hill-Robertson interference cannot result in the observed excess of LDnonsyn. Generally, Hill-Robertson interference decreases LDnonsyn, especially under low recombination rate (Hill and Robertson, 1966; Comeron et al., 2008; Garcia and Lohmueller, 2021). We discuss this in Appendix 2 (Supplementary Note 2 in the old version of the manuscript) and reproduce the effect in simulations.
While the authors argue that balancing selection is needed to account for patterns of haplotype variation they see, widespread balancing selection may not be required in this setting, and soft or partial selective sweeps (either on single mutations or sets of mutations) can also lead to patterns of diversity where a small number of haplotypes are each at appreciable frequency.
Although partial sweeps can indeed elevate LD in the linked locus, they aren’t expected to cause the excess of LDnonsyn observed in the haploblocks. In order to show this, we now simulated partial sweeps with and without epistasis. In the hard sweep model, a new beneficial mutation (s=0.5) was introduced in the population. In the soft sweep model, the beneficial mutation was picked from standing variation: selection coefficient of an initially neutral variant with frequency > 5% was changed to 0.5. In both cases, simulations were stopped when beneficial mutation achieves frequency 0.5. Both hard and soft partial sweeps increase LD as compared to simulations without sweeps (Figure R1A,B below). However, even in the presence of pairwise epistasis they don’t result in LDnonsyn > LDsyn (Figure R1C,D).
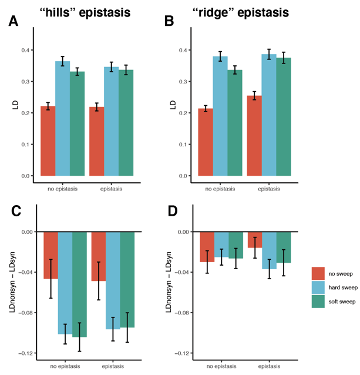
Figure R1. Patterns of LD in simulations with partial selective sweeps. Errorbars show the 95% confidence intervals obtained in 100 simulations. Simulation parameters and epistasis models are the same as described in Figure 3 - figure supplement 6.
Additionally, sweeps are expected to decrease nucleotide diversity in the linked region. However, nucleotide diversity within haploblocks observed in S. commune populations isn’t lower than in the non-haploblocks regions (Figure R2), arguing that the observed patterns can’t be caused by selective sweeps.
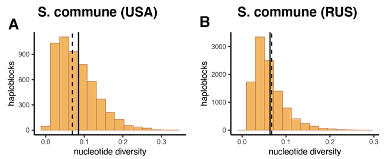
Figure R2. Nucleotide diversity in haploblocks in S. commune populations. Histograms show nucleotide diversity within haploblocks, solid black line shows the average nucleotide diversity in haploblocks. Dashed line shows the average nucleotide diversity in the non-haploblock regions.
There is also a tension between arguing that balancing selection is widespread and that shared SNPs across populations are expected to arise through recurrent mutation, as balancing selection is known to preserve haplotypes over long evolutionary times. In that section of the discussion especially, I had difficulty following the logic, and some statements are presented more definitively than might be warranted.
Although we find that balancing selection (either negative frequency-dependent selection or associative overdominance) maintains haploblocks for a long time within S. commune populations, haploblocks aren’t conserved between the two populations, as mentioned in the manuscript. Perhaps this is because balancing selection has had ample time to change on such large evolutionary scales (genetic difference between two S. commune populations is > 0.3 dS), making the fraction of identical by descent polymorphisms in the two populations low. Therefore, the SNPs that are shared between populations most probably arise by recurrent mutations, rather than descending from the ancestral population. We now clarify this in the main text.
Meanwhile, correlation of LDs between such shared SNPs in the two populations within genes indicates shared epistatic constraints between these populations. Such correlation is seen not because pairs of SNPs are maintained from the ancestral S. commune population, but because epistatic pairs are more likely to be under high LD in both modern populations.
- The validations through simulation are somewhat meagre, and I am not convinced that the simulations cover the appropriate parameter regimes. With a population size of 1000, this represents a severe down-scaling of population size and up-scaling of mutation, selection, and recombination rates (if > 0), and it's unclear if such aggressive scaling puts the simulations in an interference/interaction regime far from the true populations.
Scaling was performed according to SLiM3.0 manual in order to impove calculation time for simulations of highly diverse populations. To address the Reviewer’s concern, we now also check that this approach gives the same results as scaling of N instead of μ, as long as we scale selection coefficient s to maintain Ns and simulate for 100N generations to achieve mutation-selection equilibrium. This is indeed the case for 4Nμ up to 0.05 (Figure R3). We didn’t perform simulations for larger 4Nμ because of extremely long calculation time for large N.
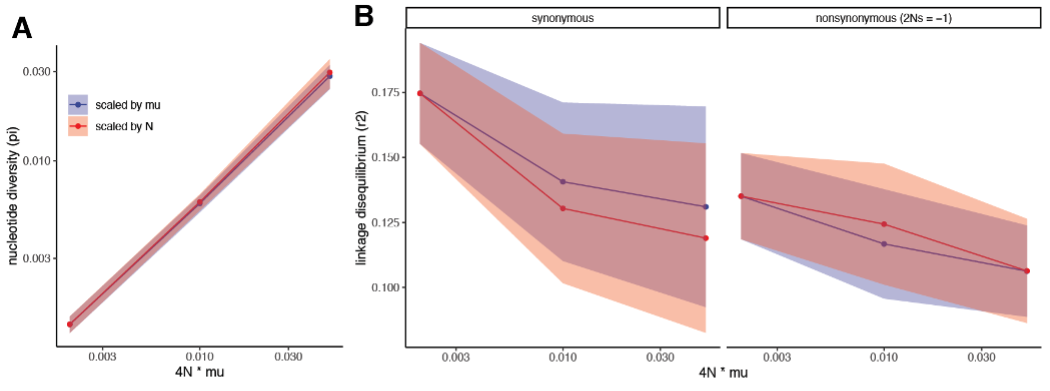
Figure R3. Simulations of populations with varying nucleotide diversity scaled by population size or mutation rate. (A) nucleotide diversity, (B) linkage disequilibrium for synonymous (s = 0) and nonsynonymous (2Ns = -1) polymorphisms. In simulations with scaled population size, mutation rate μ = 5e-7 and N is scaled to achieve 4Nμ equal to 0.002, 0.01 and 0.05. In simulations with scaled mutation rate, N = 1000 and μ is scaled accordingly. Simulations are performed for 100N generations. Filled areas show 95% confidence intervals calculated for 50 simulations with 4Nμ = 0.05; 250 simulations with 4Nμ = 0.01 and 1000 simulations with 4Nμ = 0.002.
A selection coefficient of -0.01 also implies 2Ns = -20, whereas Hill-Robertson interference is most pronounced between mutations with 2Ns ~ -1.
We performed additional simulations of evolution in a highly polymorphic population (4Nμ = 0.2) with nonsynonymous mutations under selection coefficient -5e-4 (2Ns = -1) and varying recombination rate. Consistent with the studies showing that the Hill-Robertson interference results in repulsion of deleterious variants (Hill and Robertson, 1966; Comeron et al., 2008; Garcia and Lohmueller, 2021), in our simulations, LDnonsyn is lower that LDsyn for all recombination rates (Appendix 2 - figure 4). We now append these results to Appendix 2.
- Large portions of the genome (8.4 and 15.9%, depending on the population) are covered by haploblocks, which are originally detected as genomic windows with elevated LD among SNPs. It's therefore unsurprising that haploblocks identified as high-LD outliers have elevated LD compared to other regions of the genome, and the discussion about the importance of haploblocks seemed a bit circular.
Haploblocks are surprising in two ways. Firstly, the existence of haploblocks by itself is indicative of balancing selection allowing two divergent haplotypes to persist within the population for a long time. Secondly, the strongest excess of LDnonsyn over LDsyn is oberved in genes with high LD, i.e. the ones partially or fully falling within haploblock regions (Figure 3). Positive correlation of LD and excess of LDnonsyn indicates that epistasis is more efficient in regions of high LD (haploblocks), so that the strong attraction between nonsynonymous variants observed in S. commune results from interaction between epistasis and balancing selection. We now reformulated the corresponding results section to make this clearer. We also discuss the interaction between balancing selection and epistasis in the discussion section of the manuscript.
- Finally, the authors observe a positive correlation between Pn/Ps and LD between both synonymous and nonsynonymous mutations. This result is intriguing and should be discussed, but the authors do not comment on this result in the Discussion.
Positive correlation between pn/ps, LD and the excess of LDnonsyn can be caused by multiple mechanisms, such as positive epistasis weakening the action of negative selection on nonsynonymous variants, or differences in the efficiacy of epistatic and non-epistatic selection for alleles under different allele frequency or local recombination rate. We now add the discussion on the interaction between pn/ps, LD and the excess of LDnonyn to the corresponding Results section.
-
Evaluation Summary:
Stolyarova et al. investigate a highly polymorphic species, the fungus Schizophyllum commune, finding that compared to synonymous mutations, levels of linkage disequilibrium between nonsynonymous mutations are higher within genes than between genes. The authors propose this observation may be explained by compensatory interactions between nonsynonymous alleles, pointing to the presence of positive epistasis. This paper should be of interest to population geneticists and evolutionary biologists studying the role of natural selection.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. The reviewers remained anonymous to the authors.)
-
Reviewer #1 (Public Review):
Two important goals in evolutionary biology are (i) to understand why different species exhibit different levels of genetic diversity and (ii) in each species, what is the evolutionary nature of genetic variants. Are genetic variants mostly neutral, deleterious, or advantageous? In their study, Stolyarova et al. looked at one of the most polymorphic species known, the fungus Schizophyllum commune. They found that in this hyperpolymorphic species, the evolutionary forces that govern and structure genetic variation can be very different compared to less polymorphic species, including humans and flies. Specifically, the authors find that a process known as positive epistasis is quantitatively abundant among genetic variants that alter proteins in S. commune. Positive epistasis happens when a combination of …
Reviewer #1 (Public Review):
Two important goals in evolutionary biology are (i) to understand why different species exhibit different levels of genetic diversity and (ii) in each species, what is the evolutionary nature of genetic variants. Are genetic variants mostly neutral, deleterious, or advantageous? In their study, Stolyarova et al. looked at one of the most polymorphic species known, the fungus Schizophyllum commune. They found that in this hyperpolymorphic species, the evolutionary forces that govern and structure genetic variation can be very different compared to less polymorphic species, including humans and flies. Specifically, the authors find that a process known as positive epistasis is quantitatively abundant among genetic variants that alter proteins in S. commune. Positive epistasis happens when a combination of multiple genetic variants is advantageous for the individuals that carry them, even though each isolated variant in the combination is not advantageous or even detrimental on its own. The authors explain that this happens frequently in their hyperpolymorphic species because the very high polymorphism level makes it very likely that the genetic variants will by chance occur together in the same individuals. In less polymorphic species, the variants that are advantageous in combination may have to wait for each other to occur for too long, for the combination to ever happen often enough in the first place.
Overall I had a great time reading the manuscript, and I feel that my understanding of evolution has been advanced on a fundamental level after reading it. However part of the reason why I enjoyed it was having to fill the gaps, answer the riddles left unanswered in the story by the authors.
Strengths:
The model, both extremely polymorphic and amenable to haploid cultures, is ideal to address the questions asked.
The study potentially represents a very important conceptual advance on the way to better understand genetic variation in general.
The interpretations made by the authors of their data are likely the correct ones to make, even though more definitive answers will likely only come from the sequencing of a much larger number of haplotypes, which cannot reasonably be asked of the authors at this point.
Weaknesses:
The manuscript does not provide enough information to judge if the synonymous controls that are compared to the nonsynonymous variants are fully adequate. Specifically, I have one concern that the Site Frequency Spectrum (SFS) of the synonymous variants at MAF>0.05 may be very different compared to the SFS of nonsynonymous variants at MAF>0.05. I focus on this because the authors mention page 5 line 3: "The excess of LDnonsyn over LDsyn corresponds to the attraction between rare alleles at nonsynonymous sites". First, it is unclear from this or from the figures at this point in the manuscript what the authors mean by rare alleles, among those alleles at MAF>0.05. This needs to be detailed quantitatively much more carefully. Second, and most importantly, this raises the question of whether or not the synonymous controls have a SFS with many less rare (but with MAF>0.05) alleles, as one may expect if they are under less purifying selection than nonsynonymous variants. This then raises the question of whether or not the synonymous control conducted by the author is adequate, or if the authors need to explicitly match the synonymous control in terms of SFS for MAF>0.05 in addition to the distance matching already done.
The manuscript is far too succinct on several occasions, where observations or interpretations need to be much more detailed and explained.
-
Reviewer #2 (Public Review):
Stolyarova et al. used a highly polymorphic species, Schizophyllum commune, to explore patterns of LD between nonsynonymous and synonymous mutations within protein-coding genes. LD is informative about interference and interactions between selected loci, with compensatory mutations expected to be in strong positive LD. The benefit of studying this fungal species with large diversity (with pi > 0.1) is that populations are able to explore relatively large regions of the fitness landscape, and chances increase that sets of epistatically interacting mutations segregate at the same time.
This study finds strong positive LD between pairs of nonsynonymous mutations within, but not between genes, compared to pairs synonymous variants. Further, the authors show that high LD is prevalent among pairs of mutations at …
Reviewer #2 (Public Review):
Stolyarova et al. used a highly polymorphic species, Schizophyllum commune, to explore patterns of LD between nonsynonymous and synonymous mutations within protein-coding genes. LD is informative about interference and interactions between selected loci, with compensatory mutations expected to be in strong positive LD. The benefit of studying this fungal species with large diversity (with pi > 0.1) is that populations are able to explore relatively large regions of the fitness landscape, and chances increase that sets of epistatically interacting mutations segregate at the same time.
This study finds strong positive LD between pairs of nonsynonymous mutations within, but not between genes, compared to pairs synonymous variants. Further, the authors show that high LD is prevalent among pairs of mutations at amino acid sites that interact within the protein. This result is consistent with pairs or sets of compensatory nonsynonymous mutations cosegregating within protein-coding genes.
The conclusions of this paper are largely supported by the data, with some caveats, listed below.
1. With such large pairwise diversity, there are bound to be many deleterious variants segregating at once, and the large levels of interference between them will make selection much less efficient at purging deleterious variants. While the authors argue that balancing selection is needed to account for patterns of haplotype variation they see, widespread balancing selection may not be required in this setting, and soft or partial selective sweeps (either on single mutations or sets of mutations) can also lead to patterns of diversity where a small number of haplotypes are each at appreciable frequency.
There is also a tension between arguing that balancing selection is widespread and that shared SNPs across populations are expected to arise through recurrent mutation, as balancing selection is known to preserve haplotypes over long evolutionary times. In that section of the discussion especially, I had difficulty following the logic, and some statements are presented more definitively than might be warranted.
2. The validations through simulation are somewhat meagre, and I am not convinced that the simulations cover the appropriate parameter regimes. With a population size of 1000, this represents a severe down-scaling of population size and up-scaling of mutation, selection, and recombination rates (if > 0), and it's unclear if such aggressive scaling puts the simulations in an interference/interaction regime far from the true populations. A selection coefficient of -0.01 also implies 2Ns = -20, whereas Hill-Robertson interference is most pronounced between mutations with 2Ns ~ -1.
3. Large portions of the genome (8.4 and 15.9%, depending on the population) are covered by haploblocks, which are originally detected as genomic windows with elevated LD among SNPs. It's therefore unsurprising that haploblocks identified as high-LD outliers have elevated LD compared to other regions of the genome, and the discussion about the importance of haploblocks seemed a bit circular.
4. Finally, the authors observe a positive correlation between Pn/Ps and LD between both synonymous and nonsynonymous mutations. This result is intriguing and should be discussed, but the authors do not comment on this result in the Discussion.
-
