Cell-surface tethered promiscuous biotinylators enable comparative small-scale surface proteomic analysis of human extracellular vesicles and cells
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
This report describes a new technique to detect the surface proteome of normal and myc-transformed cells in relation to extracellular vesicles from the same cells. The data obtained from this comparison may be useful in evaluating cell surface and extracellular vesicle marker proteins that may be of diagnostic value. The article could possibly be more interesting if the actual proteomic results of control vs Myc and cells vs extracellular vesicles were more extensively exploited.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 and Reviewer #2 agreed to share their names with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Characterization of cell surface proteome differences between cancer and healthy cells is a valuable approach for the identification of novel diagnostic and therapeutic targets. However, selective sampling of surface proteins for proteomics requires large samples (>10e6 cells) and long labeling times. These limitations preclude analysis of material-limited biological samples or the capture of rapid surface proteomic changes. Here, we present two labeling approaches to tether exogenous peroxidases (APEX2 and HRP) directly to cells, enabling rapid, small-scale cell surface biotinylation without the need to engineer cells. We used a novel lipidated DNA-tethered APEX2 (DNA-APEX2), which upon addition to cells promoted cell agnostic membrane-proximal labeling. Alternatively, we employed horseradish peroxidase (HRP) fused to the glycan-binding domain of wheat germ agglutinin (WGA-HRP). This approach yielded a rapid and commercially inexpensive means to directly label cells containing common N-Acetylglucosamine (GlcNAc) and sialic acid glycans on their surface. The facile WGA-HRP method permitted high surface coverage of cellular samples and enabled the first comparative surface proteome characterization of cells and cell-derived small extracellular vesicles (EVs), leading to the robust quantification of 953 cell and EV surface annotated proteins. We identified a newly recognized subset of EV-enriched markers, as well as proteins that are uniquely upregulated on Myc oncogene-transformed prostate cancer EVs. These two cell-tethered enzyme surface biotinylation approaches are highly advantageous for rapidly and directly labeling surface proteins across a range of material-limited sample types.
Article activity feed
-

-

Author Response:
Reviewer #1 (Public Review):
Cell surface proteins are of vital interest in the functions and interactions of cells and their neighbors. In addition, cells manufacture and secrete small membrane vesicles that appear to represent a subset of the cell surface protein composition.
Various techniques have been developed to allow the molecular definition of many cell surface proteins but most rely on the special chemistry of amino acid residues in exposed on the parts of membrane proteins exposed to the cell exterior.
In this report Kirkemo et al. have devised a method that more comprehensively samples the cell surface protein composition by relying on the membrane insertion or protein glycan adhesion of an enzyme that attaches a biotin group to a nearest neighbor cellular protein. The result is a more complex set of …
Author Response:
Reviewer #1 (Public Review):
Cell surface proteins are of vital interest in the functions and interactions of cells and their neighbors. In addition, cells manufacture and secrete small membrane vesicles that appear to represent a subset of the cell surface protein composition.
Various techniques have been developed to allow the molecular definition of many cell surface proteins but most rely on the special chemistry of amino acid residues in exposed on the parts of membrane proteins exposed to the cell exterior.
In this report Kirkemo et al. have devised a method that more comprehensively samples the cell surface protein composition by relying on the membrane insertion or protein glycan adhesion of an enzyme that attaches a biotin group to a nearest neighbor cellular protein. The result is a more complex set of proteins and distinctive differences between normal and a myc oncogene tumor cells and their secreted extracellular vesicle counterparts. These results may be applied to the identification of unique cell surface determinants in tumor cells that could be targets for immune or drug therapy. The results may be strengthened by a more though evaluation of the different EV membrane species represented in the broad collection of EVs used in this investigation.
We thank the reviewer for recognizing the importance of the work outlined in the manuscript. We have addressed the necessary improvements in the essential revisions section above.
Reviewer #2 (Public Review):
This paper describes two methods for labeling cell-surface proteins. Both methods involve tethering an enzyme to the membrane surface to probe the proteins present on cells and exosomes. Two different enzyme constructs are used: a single strand lipidated DNA inserted into the membrane that enables binding of an enzyme conjugated to a complementary DNA strand (DNA-APEX2) or a glycan-targeting binding group conjugated to horseradish peroxidase (WGA-HRP). Both tethered enzymes label proteins on the cell surface using a biotin substrate via a radical mechanism. The method provides significantly enhanced labeling efficiency and is much faster than traditional chemical labeling methods and methods that employ soluble enzymes. The authors comprehensively analyze the labeled proteins using mass spectrometry and find multiple proteins that were previously undetectable with chemical methods and soluble enzymes. Furthermore, they compare the labeling of both cells and the exosomes that are formed from the cells and characterize both up- and down-regulated proteins related to cancer development that may provide a mechanistic underpinning.
Overall, the method is novel and should enable the discovery of many low-abundance cell-surface proteins through more efficient labeling. The DNA-APEX2 method will only be accessible to more sophisticated laboratories that can carry out the protocols but the WGA-HRP method employs a readily available commercial product and give equivalent, perhaps even better, results. In addition, the method cannot discriminate between proteins that are genuinely expressed on the cell from those that are non-specifically bound to the cell surface.
The authors describe the approach and identify two unique proteins on the surface of prostate cell lines.
Strengths:
Good introduction with appropriate citations of relevant literature Much higher labeling efficiency and faster than chemical methods and soluble enzyme methods. Ability to detect low-abundance proteins, not accessible from previous labeling methods.
Weaknesses: The DNA-APEX2 method requires specialized reagents and protocols that are much more challenging for a typical laboratory to carry out than conventional chemical labeling methods.
The claims and findings are sound. The finding of novel proteins and the quantitative measurement of protein up- and down-regulation are important. The concern about non-specifically bound proteins could be addressed by looking at whether the detected proteins have a transmembrane region that would enable them to localize in the cell membrane.
We thank the reviewer for recognizing the strengths and importance of this work. We also thank the reviewer for mentioning the issue of non-specifically bound proteins. As addressed above in the essential revisions sections, we believe that any low affinity, non-specific binding proteins are likely removed in the multiple wash/centrifugation steps on cells or the multiple centrifugation steps and sucrose gradient purification on EVs. Given the likelihood for removal of non-specific binders, we believe that the secreted proteins identified are likely high affinity interactions and their differential expression on either cells or EVs play an important part in the downstream biology of both sample types. However, the previous data presentation did not clarify which proteins pertained to the transmembrane plasma membrane proteome versus secreted protein forms. For further clarity in the data presentation (Figure 3D, 4D, 5D), we have bolded proteins that are also found in the SURFY database that only includes surface annotated proteins with a predicted transmembrane domain (Bausch-Fluck et al., The in silico human surfaceome. PNAS. 2018). We have also italicized proteins that are annotated to be secreted from the cell to the extracellular space (Uniprot classification). We have updated the text and caption as shown below:
New Figure 3:
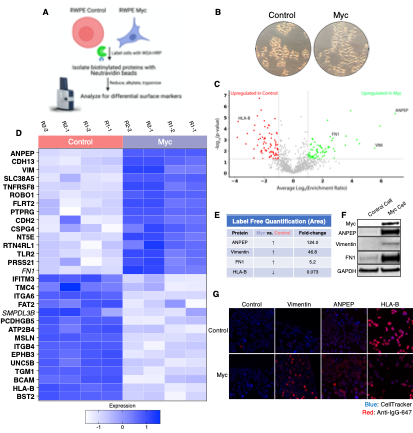
Figure 3. WGA-HRP identifies a number of enriched markers on Myc-driven prostate cancer cells. (A) Overall scheme for biotin labeling, and label-free quantification (LFQ) by LC-MS/MS for RWPE-1 Control and Myc over-expression cells. (B) Microscopy image depicting morphological differences between RWPE-1 Control and RWPE-1 Myc cells after 3 days in culture. (C) Volcano plot depicting the LFQ comparison of RWPE-1 Control and Myc labeled cells. Red labels indicate upregulation in the RWPE-1 Control cells over Myc cells and green labels indicate upregulation in the RWPE-1 Myc cells over Control cells. All colored proteins are 2-fold enriched in either dataset between four replicates (two technical, two biological, p<0.05). (D) Heatmap of the 15 most upregulated transmembrane (bold) or secreted (italics) proteins in RWPE-1 Control and Myc cells. Scale indicates intensity, defined as (LFQ Area - Mean LFQ Area)/standard deviation. Extracellular proteins with annotated transmembrane domains are bolded and annotated secreted proteins are italicized. (E) Table indicating fold-change of most differentially regulated proteins by LC-MS/MS for RWPE-1 Control and Myc cells. (F) Upregulated proteins in RWPE-1 Myc cells (Myc, ANPEP, Vimentin, and FN1) are confirmed by western blot. (G) Upregulated surface proteins in RWPE-1 Myc cells (Vimentin, ANPEP, FN1) are detected by immunofluorescence microscopy. The downregulated protein HLA-B by Myc over-expression was also detected by immunofluorescence microscopy. All western blot images and microscopy images are representative of two biological replicates. Mass spectrometry data is based on two biological and two technical replicates (N = 4).
New Figure 4:
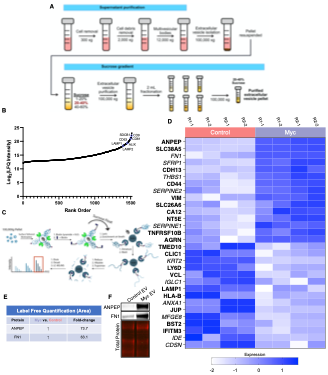
Figure 4. WGA-HRP identifies a number of enriched markers on Myc-driven prostate cancer EVs. (A) Workflow for small EV isolation from cultured cells. (B) Labeled proteins indicating canonical exosome markers (ExoCarta Top 100 List) detected after performing label-free quantification (LFQ) from whole EV lysate. The proteins are graphed from least abundant to most abundant. (C) Workflow of exosome labeling and preparation for mass spectrometry. (D) Heatmap of the 15 most upregulated proteins in RWPE-1 Control or Myc EVs. Scale indicates intensity, defined as (LFQ Area - Mean LFQ Area)/SD. Extracellular proteins with annotated transmembrane domains are bolded and annotated secreted proteins are italicized. (E) Table indicating fold-change of most differentially regulated proteins by LC-MS/MS for RWPE-1 Control and Myc cells. (F) Upregulated proteins in RWPE-1 Myc EVs (ANPEP and FN1) are confirmed by western blot. Mass spectrometry data is based on two biological and two technical replicates (N = 4). Due to limited sample yield, one replicate was performed for the EV western blot.
New Figure 5:
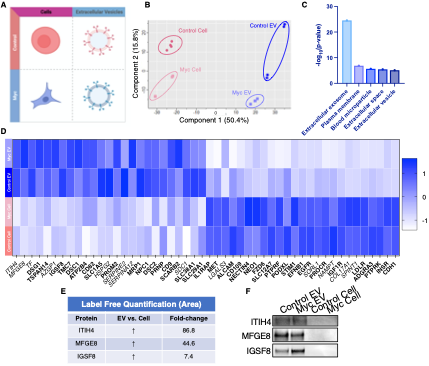
Figure 5. WGA-HRP identifies a number of EV-specific markers that are present regardless of oncogene status. (A) Matrix depicting samples analyzed during LFQ comparison--Control and Myc cells, as well as Control and Myc EVs. (B) Principle component analysis (PCA) of all four groups analyzed by LFQ. Component 1 (50.4%) and component 2 (15.8%) are depicted. (C) Functional annotation clustering was performed using DAVID Bioinformatics Resource 6.8 to classify the major constituents of component 1 in PCA analysis. (D) Heatmap of the 25 most upregulated proteins in RWPE-1 cells or EVs. Proteins are listed in decreasing order of expression with the most highly expressed proteins in EVs on the far left and the most highly expressed proteins in cells on the far right. Scale indicates intensity, defined as (LFQ Area - Mean LFQ Area)/SD. Extracellular proteins with annotated transmembrane domains are bolded and annotated secreted proteins are italicized. (E) Table indicating fold-change of most differentially regulated proteins by LC-MS/MS for RWPE-1 EVs compared to parent cells. (F) Western blot showing the EV specific marker ITIH4, IGSF8, and MFGE8.Mass spectrometry data is based on two biological and two technical replicates (N = 4). Due to limited sample yield, one replicate was performed for the EV western blot.
Authors mention time-sensitive changes but it is unclear how this method would enable one to obtain this kind of data. How would this be accomplished? The statement "Due to the rapid nature of peroxidase enzymes (1-2 min), our approaches enable kinetic experiments to capture rapid changes, such as binding, internalization, and shuttling events." Yes, it is faster, but not sure I can think of an experiment that would enable one to capture such events.
We thank the reviewer for this comment and giving us an opportunity to elaborate on the types of experiments enabled by this new method. A previous study (Y, Li et al. Rapid Enzyme-Mediated Biotinylation for Cell Surface Proteome Profiling. Anal. Chem. 2021) showed that labeling the cell surface with soluble HRP allowed the researchers to detect immediate surface protein changes in response to insulin treatment. They demonstrated differential surfaceome profiling changes at 5 minutes vs 2 hours following treatment with insulin. Only methods utilizing these rapid labeling enzymes could allow for this type of resolution. A few other biological settings that experience rapid cell surface changes are: response to drug treatment, T-cell activation and synapse formation (S, Valitutti, et al. The space and time frames of T cell activation at the immunological synapse. FEBS Letters. 2010) and GPCR activation (T, Gupte et al. Minute-scale persistence of a GPCR conformation state triggered by non-cognate G protein interactions primes signaling. Nat. Commun. 2019). We also believe the method would be useful for post-translational processes where proteins are rapidly shuttling to the cell surface. We have updated the discussion to elaborate on these types of experiments.
"Due to the fast kinetics of peroxidase enzymes (1-2 min), our approaches could enable kinetic experiments to capture rapid post-translational trafficking of surfaces proteins, such as response to insulin, certain drug treatments, T-cell activation and synapse formation, and GPCR activation."
The authors do not have any way to differentiate between proteins expressed by cells and presented on their membranes from proteins that non-specifically bind to the membrane surface. Non-specific binding (NSB) is not addressed. Proteins can non-specifically bind to the cell or EV surface. The results are obtained by comparisons (cells vs exosomes, controls vs cancer cells), which is fine because it means that what is being measured is differentially expressed, so even NSB proteins may be up- and down-regulated. But the proteins identified need to be confirmed. For example, are all the proteins being detected transmembrane proteins that are known to be associated with the membrane?
As mentioned above, we utilized the most rigorous informatics analysis available (Uniprot and SURFY) to annotate the proteins we find as having a signal sequence and/or TM domain. Data shown in heatmaps are based off of significance (p < 0.05) across all four replicates, which supports that any secreted proteins present are likely due to actual biological differences between oncogenic status and/or sample origin (i.e. EV vs cell). We have addressed this point in a previous comment above.
The term "extracellular vesicles" (EVs) might be more appropriate than "exosomes" to describe the studied preparation.
As we describe above in response to earlier comments, we have systematically changed from using exosomes to small extracellular vesicles and better defined the isolation procedure that we used in the methods section.
Reviewer #3 (Public Review):
The article by Kirkemo et al explores approaches to analyse the surface proteome of cells or cell-derived extracellular vesicles (EVs, called here exosomes, but the more generic term "extracellular vesicles" would be more appropriate because the used procedure leads to co-isolation of vesicles of different origin), using tools to tether proximity-biotinylation enzymes to membranes. The authors determine the best conditions for surface labeling of cells, and demonstrate that tethering the enzymes (APEX or HRP) increases the number of proteins detected by mass-spectrometry. They further use one of the two approaches (where HRP binds to glycans), to analyse the biotinylated proteome of two variants of a prostate cancer cell line, and the corresponding EVs. The approaches are interesting, but their benefit for analysis of cells or EVs is not very strongly supported by the data.
First, the authors honestly show (fig2-suppl figures) that only 35% of the proteins identified after biotinylation with their preferred tool actually correspond to annotated surface proteins. This is only slightly better than results obtained with a non-tethered sulfo-NHS-approach (30%).
We thank the reviewer for this comment. The reason we utilize membrane protein enrichment methods is that membrane protein abundance is low compared to cytosolic proteins and their identification can be overwhelmed by cytosolic contaminants. Nonetheless, despite our best efforts to limit labeling to the membrane proteins, cytosolic proteins can carry over. Thus, we utilize informatics methods to identify the proteins that are annotated to be membrane associated. The Uniprot GOCC (Gene Ontology Cellular Component) Plasma Membrane database is the most inclusive of membrane proteins only requiring they contain either a signal sequence, transmembrane domain, GPI anchor or other membrane associated motifs yielding a total of 5,746 proteins. This will include organelle membrane proteins. It is known that proteins can traffic from the internal organelles to the cell surface so these can be bonified cell surface proteins too. To increase the informatics stringency for membrane proteins we have now applied a new database aggregated from work by the Wollscheid lab, called SURFY (Bausch-Fluck et al., The in silico human surfaceome. PNAS. 2018). This is a machine learning method trained on 735 high confidence membrane proteins from the Cell Surface Protein Atlas (CSPA). SURFY predicts a total of 2,886 cell surface proteins. When we filter our data using SURFY for proteins, peptides and label free quantitation (LFQ) area for three methods, we find that the difference between NHS-Biotin and WGA-HRP expands considerably (see new Figure 3-Supplemental Figure 1 below). We observe these differences when the datasets are searched with either the GOCC Plasma Membrane database or the entire human Uniprot database. The difference is especially large for LFQ analysis, which quantitatively scores peptide intensity as opposed to simply count the number hits as for protein and peptide analysis. Cytosolic carry over is the major disadvantage of NHS-Biotin, which suppresses signal strength and is reflected in the lower LFQ values (24% for NHS-biotin compared to 40% for WGA-HRP). We have updated the main text and supplemental figure below:
"Both WGA-HRP and biocytin hydrazide had similar levels of cell surface enrichment on the peptide and protein level when cross-referenced with the SURFY curated database for extracellular surface proteins with a predicted transmembrane domain (Figure 3 - Figure supplement 1A). Sulfo-NHS-LC-LC-biotin and whole cell lysis returned the lowest percentage of cell surface enrichment, suggesting a larger portion of the total sulfo-NHS-LC-LC-biotin protein identifications were of intracellular origin, despite the use of the cell-impermeable format. These same enrichment levels were seen when the datasets were searched with the curated GOCC-PM database, as well as the Uniprot entire human proteome database (Figure 3 - Figure supplement 1B). Importantly, of the proteins quantified across all four conditions, biocytin hydrazide and WGA-HRP returned higher overall intensity values for SURFY-specified proteins than either sulfo-NHS-LC-LC-biotin or whole cell lysis. Importantly, although biocytin hydrazide shows slightly higher cell surface enrichment compared to WGA-HRP, we were unable to perform the comparative analysis at 500,000 cells--instead requiring 1.5 million--as the protocol yielded too few cells for analysis."
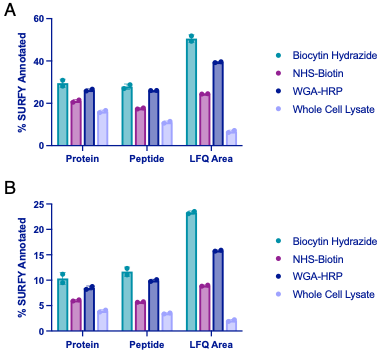
Figure 3-Figure Supplement 1. Comparison of surface enrichment between replicates for different mass spectrometry methods. (A) The top three methods (NHS-Biotin, Biocytin Hydrazide, and WGA-HRP) were compared for their ability to enrich cell surface proteins on 1.5 M RWPE-1 Control cells by LC-MS/MS after being searched with the Uniprot GOCC Plasma Membrane database. Shown are enrichment levels on the protein, peptide, and average MS1 intensity of top three peptides (LFQ area) levels. (B) The top three methods (NHS-Biotin, Biocytin Hydrazide, and WGA-HRP) were compared for their ability to enrich cell surface proteins on 1.5 M RWPE-1 Control cells by LC-MS/MS after being searched with the entire human Uniprot database. Shown are enrichment levels on the protein, peptide, and average MS1 intensity of top three peptides (LFQ area) levels. Proteins or peptides detected from cell surface annotated proteins (determined by the SURFY database) were divided by the total number of proteins or peptides detected. LFQ areas corresponding to cell surface annotated proteins (SURFY) were divided by the total area sum intensity for each sample. The corresponding percentages for two biological replicates were plotted.
There are additional advantages to WGA-HRP over NHS-biotin. These include: (i) labeling time is 2 min versus 30 min, which would afford higher kinetic resolution as needed, and (ii) the NHS-biotin labels lysines, which hinders tryptic cleavage and downstream peptide analysis, whereas the WGA-HRP labels tyrosines, eliminating impacts on tryptic patterns. WGA-HRP is slightly below biocytin hydrazide in peptide and protein ID and somewhat more by LFQ. However, there are significant advantages over biocytin hydrazide: (i) sample size for WGA-HRP can be reduced a factor of 3-5 because of cell loss during the multiple washing steps after periodate oxidation and hydrazide labeling, (ii) the time of labeling is dramatically reduced from 3 hr for hydrazide to 2 min for WGA-HRP, and (iii) the HRP enzyme has a large labeling diameter (20-40 nm, but also reported up to 200 nm) and can label non-glycosylated membrane proteins as opposed to biocytin hydrazide that only labels glycosylated proteins. The hydrazide method is the current standard for membrane protein enrichment, and we feel that the WGA-HRP will compete especially when cell sample size is limited or requires special handling. In the case of EVs, we were not able to perform hydrazide labeling due to the two-step process and small sample size.
Indeed the list of identified proteins in figures 4 and 5 include several proteins whose expected subcellular location is internal, not surface exposed, and whose location in EVs should also be inside (non-exhaustively: SDCBP = syntenin, PDCD6IP = Alix, ARRDC1, VPS37B, NUP35 = nucleopore protein)…
We thank the reviewer for this comment. We have elaborated on this point in a number of response paragraphs above. The proteins that the reviewer points out are annotated as “plasma membrane” in the very inclusive GOCC plasma membrane database. However, this means that they may also spend time in other locations in the cell or reside on organelle membranes. We have done further analysis to remove any intracellular membrane residing proteins that are included in the GOCC plasma membrane database, including the five proteins mentioned above. We also have further highlighted proteins that appear in the SURFY database, as discussed above and in our response to Reviewer 2’s comment. To increase stringency, we have bolded proteins that are found in the more selective SURFY database and italicized secreted proteins. Due to our new analysis and data presentation, it is more clear which markers are bona fide extracellular resident membrane proteins. We have updated the Figures and Figure legends as mentioned above, as well as added this statement in the Data Processing and Analysis methods:
"Additionally, to not miss any key surface markers such as secreted proteins or anchored proteins without a transmembrane domain, we chose to initially avoid searching with a more stringent protein list, such as the curated SURFY database. However, following the analysis, we bolded proteins found in the SURFY database and italicized proteins known to be secreted (Uniprot)."
The membrane proteins identified as different between the control and Myc-overexpressing cells or their EVs, would have been identified as well by a regular proteomic analysis.
To directly compare surfaceomes of EVs to cells, we are compelled to use the same proteomic method. For parental cell surfaceomic analysis, a membrane enrichment method is required due to the high levels of cytosolic proteins that swamp out signal from membrane proteins. Although EVs have a higher proportion of membrane to cytosol, whole EV proteomics would still have significant cytosolic contamination.
Second, the title highlights the benefit of the technique for small-scale samples: this is demonstrated for cells (figures 1-2), but not for EVs: no clear quantitative indication of amount of material used is provided for EV samples. Furthermore, no comparison with other biotinylation technics such as sulfo-NHS is provided for EVs/exosomes. Therefore, it is difficult to infer the benefit of this technic applied to the analysis of EVs/exosomes.
We appreciate the reviewer for this comment. We have updated the methods as mentioned above in our response to the Essential Revisions. In brief, the yield of EVs post-sucrose gradient isolation was 3-5 µg of protein from 16x15 cm2 plates of cells, totaling 240 mL of media. Since we had previously demonstrated that our method was superior to sulfo-NHS for enriching surface proteins on cells, we proceeded to use the WGA-HRP for the EV labeling experiments.
In addition, the WGA-based tethering approach, which is the only one used for the comparative analysis of figures 4 and 5, possibly induces a bias towards identification of proteins with a particular glycan signature: a novelty would possibly have come from a comparison of this approach with the other initially evaluated, the DNA-APEX one, where tethering is induced by lipid moieties, thus should not depend on glycans. The authors may have then identified by LC-MS/MS specific glycan-associated versus non-glycan-associated proteins in the cells or EVs membranes. Also ideally, the authors should have compared the 4 combinations of the 2 enzymes (APEX and HRP) and 2 tethers (lipid-bound DNA and WGA) to identify the bias introduced by each one.
We thank the reviewer for this comment. We performed analysis to determine whether there was a bias towards Uniprot annotated “Glyco” vs “Non-Glyco” surface proteins within the SURFY database identified across the WGA-HRP, APEX2-DNA, APEX2, and HRP labeling methods. We performed this analysis by measuring the total LFQ area detected for each category (glycoprotein vs non-glycoprotein) and dividing that by the total LFQ area found across all proteins detected in the sample. We found similar normalized areas of non-glyco surface proteins between WGA-HRP and APEX2-DNA suggesting there is not a bias against non-glycosylated proteins in the WGA-HRP sample. There were slightly elevated levels of Glycoproteins in the WGA-HRP sample over APEX2-DNA. It is not surprising to us that there is little bias because the free-radicals generated by biotin-tyramide can label over tens of nanometers and thus can label not just the protein they are attached to, but neighbors also, regardless of glycosylation status. We have added this as Figure 2-Supplement 3, and amended the text in the manuscript below in purple.
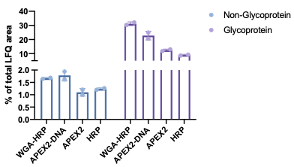
Figure 2 – Figure Supplement 3: Comparison of enrichment of Glyco- vs Non-Glyco-proteins. (A) TIC area of Uniprot annotated Glycoproteins compared to Non-Glycoproteins in the SURFY database for each labeling method compared to total TIC area. There was not a significant difference in detection of Non-Glycoproteins detected between WGA-HRP and APEX2-DNA and only a slightly higher detection of Glycoproteins in the WGA-HRP sample over APEX2-DNA.
"As the mode of tethering WGA-HRP involves GlcNAc and sialic acid glycans, we wanted to determine whether there was a bias towards Uniprot annotated 'Glycoprotein' vs 'Non-Glycoprotein' surface proteins identified across the WGA-HRP, APEX2-DNA, APEX2, and HRP labeling methods. We looked specifically looked at surface proteins founds in the SURFY database, which is the most restrictive surface database and requires that proteins have a predicted transmembrane domain (Bausch-Fluck et al., The in silico human surfaceome. PNAS. 2018). We performed this analysis by measuring the average MS1 intensity across the top three peptides (area) for SURFY glycoproteins and non-glycoproteins for each sample and dividing that by the total LFQ area found across all GOCC annotated membrane proteins detected in each sample. We found similar normalized areas of non-glyco surface proteins across all samples (Figure 2 - Figure supplement 4). If a bias existed towards glycosylated proteins in WGA-HRP compared to the glycan agnostic APEX2-DNA sample, then we would have seen a larger percentage of non-glycosylated surface proteins identified in APEX2-DNA over WGA-HRP. Due to the large labeling radius of the HRP enzyme, we find it unsurprising that the WGA-HRP method is able to capture non-glycosylated proteins on the surface to the same degree (Rees et al. Selective Proteomic Proximity Labeling Assay SPPLAT. Current Protocols in Protein Science. 2015). There is a slight increase in the area percentage of glycoproteins detected in the WGA-HRP compared to the APEX2-DNA sample but this is likely due to the fact that a greater number of surface proteins in general are detected with WGA-HRP."
As presented the article is thus an interesting technical description, which does not convince the reader of its benefit to use for further proteomic analyses of EVs or cells. Such info is of course interesting to share with other scientists as a sort of "negative" or "neutral" result. Maybe a novelty of the presented work is the differential proteome analysis of surface enriched EV/cell proteins in control versus myc-expressing cells. Such analyses of EVs from different derivatives of a tumor cell line have been performed before, for instance comparing cells with different K-Ras mutations (Demory-Beckler, Mol Cell proteomics 2013 # 23161513). However, here the authors compare also cells and EVs, and find possibly interesting discrepancies in the upregulated proteins. These results could probably be exploited more extensively. For instance, authors could give clearer info (lists) on the proteins differentially regulated in the different comparisons: in EVs from both cells, in EVs vs cells, in both cells.
We appreciate the reviewer for this critique and have updated the manuscript accordingly. We have changed the title to “Cell surface tethered promiscuous biotinylators enable small-scale comparative surface proteomic analysis of human extracellular vesicles and cells” to more accurately depict the focus of our manuscript which, as the reviewer highlighted, is that this technology allows for comparative analysis between the surfaceomes of cells vs EVs. We appreciate the fine work from the Coffey lab on whole EV analysis of KRAS transformed cells. They identified a mix of surface and cytosolic proteins that change in EVs from the transformed cells, whereas our data focuses specifically on the surfaceome differences in Myc transformed and non-transformed cells and corresponding small EVs. We believe this makes important contributions to the field as well.
To further address the reviewer’s suggestions, we additionally have significantly reorganized the figures to better display the differentially regulated proteins. We have removed the volcano plots and instead included heatmaps with the top 30 (Figure 3 and Figure 4) and top 50 (Figure 5) differentially regulated proteins across cells and EVs. We have also updated the lists of proteins in the supplemental source tables section. See responses to Reviewer 2 above for the updates to Figures 3-5. We have additionally included supplemental figures with lists of differentially upregulated proteins in the EV and Cell samples, which are shown below:
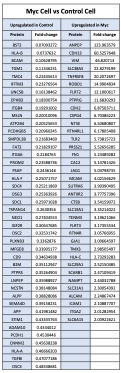
Figure 3 – Supplement 3: List of proteins comparing enriched targets (>2-fold) in Myc cells versus Control cells. Targets that were found enriched (Myc/Control) in the Control cells (left) and Myc cells (right). The fold-change between Myc cells and Control cells is listed in the column to the right of the gene name.
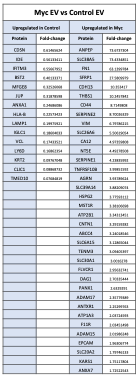
Figure 4 – Supplement 1: List of proteins comparing enriched targets (>1.5-fold) in Myc EVs versus Control EVs. Targets that were found enriched (Myc/Control) in the Control EVs (left) and Myc EVs (right). The fold-change between Myc EVs and Control EVs is listed in the column to the right of the gene name.
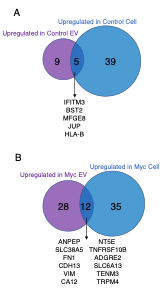
Figure 4 – Figure Supplement 2: Venn diagram comparing enriched targets (>2-fold) in Cells and EVs. (A) Targets that were found enriched in the Control EVs (purple) and Control cells (blue) when each is separately compared to Myc EVs and Myc cells, respectively. The 5 overlapping enriched targets in common between Control cells and Control EVs are listed in the center. (B) Targets that were found enriched in the Myc EVs (purple) and Myc cells (blue) when each is separately compared to Control EVs and Control cells, respectively. The 12 overlapping enriched targets in common between Myc cells and Myc EVs are listed in the center.
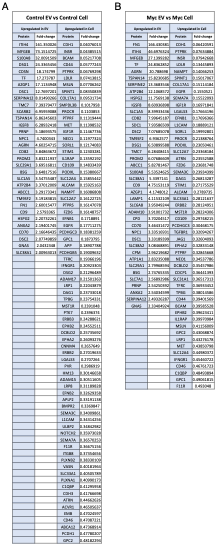
Figure 5 - Supplement 1: List of proteins comparing enriched targets (>2-fold) in Control EVs versus Control cells and Myc EVs versus Myc cells. (A)Targets that were found enriched (EV/cell) in the Control samples are listed. The fold-change values between Control EVs and Control cells are listed in the column to the right of the gene name. (B)Targets that were found enriched (EV/cell) in the Myc samples are listed. The fold-change values between Myc EVs and Myc cells are listed in the column to the right of the gene name.
-
Evaluation Summary:
This report describes a new technique to detect the surface proteome of normal and myc-transformed cells in relation to extracellular vesicles from the same cells. The data obtained from this comparison may be useful in evaluating cell surface and extracellular vesicle marker proteins that may be of diagnostic value. The article could possibly be more interesting if the actual proteomic results of control vs Myc and cells vs extracellular vesicles were more extensively exploited.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 and Reviewer #2 agreed to share their names with the authors.)
-
Reviewer #1 (Public Review):
Cell surface proteins are of vital interest in the functions and interactions of cells and their neighbors. In addition, cells manufacture and secrete small membrane vesicles that appear to represent a subset of the cell surface protein composition.
Various techniques have been developed to allow the molecular definition of many cell surface proteins but most rely on the special chemistry of amino acid residues in exposed on the parts of membrane proteins exposed to the cell exterior.
In this report Kirkemo et al. have devised a method that more comprehensively samples the cell surface protein composition by relying on the membrane insertion or protein glycan adhesion of an enzyme that attaches a biotin group to a nearest neighbor cellular protein. The result is a more complex set of proteins and distinctive …
Reviewer #1 (Public Review):
Cell surface proteins are of vital interest in the functions and interactions of cells and their neighbors. In addition, cells manufacture and secrete small membrane vesicles that appear to represent a subset of the cell surface protein composition.
Various techniques have been developed to allow the molecular definition of many cell surface proteins but most rely on the special chemistry of amino acid residues in exposed on the parts of membrane proteins exposed to the cell exterior.
In this report Kirkemo et al. have devised a method that more comprehensively samples the cell surface protein composition by relying on the membrane insertion or protein glycan adhesion of an enzyme that attaches a biotin group to a nearest neighbor cellular protein. The result is a more complex set of proteins and distinctive differences between normal and a myc oncogene tumor cells and their secreted extracellular vesicle counterparts. These results may be applied to the identification of unique cell surface determinants in tumor cells that could be targets for immune or drug therapy. The results may be strengthened by a more though evaluation of the different EV membrane species represented in the broad collection of EVs used in this investigation.
-
Reviewer #2 (Public Review):
This paper describes two methods for labeling cell-surface proteins. Both methods involve tethering an enzyme to the membrane surface to probe the proteins present on cells and exosomes. Two different enzyme constructs are used: a single strand lipidated DNA inserted into the membrane that enables binding of an enzyme conjugated to a complementary DNA strand (DNA-APEX2) or a glycan-targeting binding group conjugated to horseradish peroxidase (WGA-HRP). Both tethered enzymes label proteins on the cell surface using a biotin substrate via a radical mechanism. The method provides significantly enhanced labeling efficiency and is much faster than traditional chemical labeling methods and methods that employ soluble enzymes. The authors comprehensively analyze the labeled proteins using mass spectrometry and find …
Reviewer #2 (Public Review):
This paper describes two methods for labeling cell-surface proteins. Both methods involve tethering an enzyme to the membrane surface to probe the proteins present on cells and exosomes. Two different enzyme constructs are used: a single strand lipidated DNA inserted into the membrane that enables binding of an enzyme conjugated to a complementary DNA strand (DNA-APEX2) or a glycan-targeting binding group conjugated to horseradish peroxidase (WGA-HRP). Both tethered enzymes label proteins on the cell surface using a biotin substrate via a radical mechanism. The method provides significantly enhanced labeling efficiency and is much faster than traditional chemical labeling methods and methods that employ soluble enzymes. The authors comprehensively analyze the labeled proteins using mass spectrometry and find multiple proteins that were previously undetectable with chemical methods and soluble enzymes. Furthermore, they compare the labeling of both cells and the exosomes that are formed from the cells and characterize both up- and down-regulated proteins related to cancer development that may provide a mechanistic underpinning.
Overall, the method is novel and should enable the discovery of many low-abundance cell-surface proteins through more efficient labeling. The DNA-APEX2 method will only be accessible to more sophisticated laboratories that can carry out the protocols but the WGA-HRP method employs a readily available commercial product and give equivalent, perhaps even better, results. In addition, the method cannot discriminate between proteins that are genuinely expressed on the cell from those that are non-specifically bound to the cell surface.
The authors describe the approach and identify two unique proteins on the surface of prostate cell lines.
Strengths:
Good introduction with appropriate citations of relevant literature
Much higher labeling efficiency and faster than chemical methods and soluble enzyme methods.
Ability to detect low-abundance proteins, not accessible from previous labeling methods.Weaknesses: The DNA-APEX2 method requires specialized reagents and protocols that are much more challenging for a typical laboratory to carry out than conventional chemical labeling methods.
The claims and findings are sound. The finding of novel proteins and the quantitative measurement of protein up- and down-regulation are important. The concern about non-specifically bound proteins could be addressed by looking at whether the detected proteins have a transmembrane region that would enable them to localize in the cell membrane.
Authors mention time-sensitive changes but it is unclear how this method would enable one to obtain this kind of data. How would this be accomplished? The statement "Due to the rapid nature of peroxidase enzymes (1-2 min), our approaches enable kinetic experiments to capture rapid changes, such as binding, internalization, and shuttling events." Yes, it is faster, but not sure I can think of an experiment that would enable one to capture such events.
The authors do not have any way to differentiate between proteins expressed by cells and presented on their membranes from proteins that non-specifically bind to the membrane surface. Non-specific binding (NSB) is not addressed. Proteins can non-specifically bind to the cell or EV surface. The results are obtained by comparisons (cells vs exosomes, controls vs cancer cells), which is fine because it means that what is being measured is differentially expressed, so even NSB proteins may be up- and down-regulated. But the proteins identified need to be confirmed. For example, are all the proteins being detected transmembrane proteins that are known to be associated with the membrane?
The term "extracellular vesicles" (EVs) might be more appropriate than "exosomes" to describe the studied preparation.
-
Reviewer #3 (Public Review):
The article by Kirkemo et al explores approaches to analyse the surface proteome of cells or cell-derived extracellular vesicles (EVs, called here exosomes, but the more generic term "extracellular vesicles" would be more appropriate because the used procedure leads to co-isolation of vesicles of different origin), using tools to tether proximity-biotinylation enzymes to membranes. The authors determine the best conditions for surface labeling of cells, and demonstrate that tethering the enzymes (APEX or HRP) increases the number of proteins detected by mass-spectrometry. They further use one of the two approaches (where HRP binds to glycans), to analyse the biotinylated proteome of two variants of a prostate cancer cell line, and the corresponding EVs. The approaches are interesting, but their benefit for …
Reviewer #3 (Public Review):
The article by Kirkemo et al explores approaches to analyse the surface proteome of cells or cell-derived extracellular vesicles (EVs, called here exosomes, but the more generic term "extracellular vesicles" would be more appropriate because the used procedure leads to co-isolation of vesicles of different origin), using tools to tether proximity-biotinylation enzymes to membranes. The authors determine the best conditions for surface labeling of cells, and demonstrate that tethering the enzymes (APEX or HRP) increases the number of proteins detected by mass-spectrometry. They further use one of the two approaches (where HRP binds to glycans), to analyse the biotinylated proteome of two variants of a prostate cancer cell line, and the corresponding EVs. The approaches are interesting, but their benefit for analysis of cells or EVs is not very strongly supported by the data.
First, the authors honestly show (fig2-suppl figures) that only 35% of the proteins identified after biotinylation with their preferred tool actually correspond to annotated surface proteins. This is only slightly better than results obtained with a non-tethered sulfo-NHS-approach (30%). Indeed the list of identified proteins in figures 4 and 5 include several proteins whose expected subcellular location is internal, not surface exposed, and whose location in EVs should also be inside (non-exhaustively: SDCBP = syntenin, PDCD6IP = Alix, ARRDC1, VPS37B, NUP35 = nucleopore protein)... The membrane proteins identified as different between the control and Myc-overexpressing cells or their EVs, would have been identified as well by a regular proteomic analysis.
Second, the title highlights the benefit of the technique for small-scale samples: this is demonstrated for cells (figures 1-2), but not for EVs: no clear quantitative indication of amount of material used is provided for EV samples. Furthermore, no comparison with other biotinylation technics such as sulfo-NHS is provided for EVs/exosomes. Therefore, it is difficult to infer the benefit of this technic applied to the analysis of EVs/exosomes.
In addition, the WGA-based tethering approach, which is the only one used for the comparative analysis of figures 4 and 5, possibly induces a bias towards identification of proteins with a particular glycan signature: a novelty would possibly have come from a comparison of this approach with the other initially evaluated, the DNA-APEX one, where tethering is induced by lipid moieties, thus should not depend on glycans. The authors may have then identified by LC-MS/MS specific glycan-associated versus non-glycan-associated proteins in the cells or EVs membranes. Also ideally, the authors should have compared the 4 combinations of the 2 enzymes (APEX and HRP) and 2 tethers (lipid-bound DNA and WGA) to identify the bias introduced by each one.
As presented the article is thus an interesting technical description, which does not convince the reader of its benefit to use for further proteomic analyses of EVs or cells. Such info is of course interesting to share with other scientists as a sort of "negative" or "neutral" result.
Maybe a novelty of the presented work is the differential proteome analysis of surface enriched EV/cell proteins in control versus myc-expressing cells. Such analyses of EVs from different derivatives of a tumor cell line have been performed before, for instance comparing cells with different K-Ras mutations (Demory-Beckler, Mol Cell proteomics 2013 # 23161513). However, here the authors compare also cells and EVs, and find possibly interesting discrepancies in the upregulated proteins. These results could probably be exploited more extensively. For instance, authors could give clearer info (lists) on the proteins differentially regulated in the different comparisons: in EVs from both cells, in EVs vs cells, in both cells. -
