m6A modifications regulate intestinal immunity and rotavirus infection
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
This paper is of interest within the field of virus infection and RNA methylation. The data in the manuscript provide novel information on RNA methylation during rotavirus infection but do not fully support the conclusion because of several technical issues and experimental design.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. The reviewers remained anonymous to the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
N6-methyladenosine (m6A) is an abundant mRNA modification that affects many biological processes. Nevertheless, we have a poor understanding of how m6A levels are regulated during physiological or pathological processes such as responses to virus infections. The in vivo function of m6A in intestinal immune defense against virus infections is largely unknown. Here, we describe a novel antiviral function of m6A modification during rotavirus (RV) infection in small bowel intestinal epithelial cells (IECs). We found that rotavirus infection induced global m6A modifications on mRNA transcripts by downregulating the m6a eraser ALKBH5. Mice lacking the m6A writer enzymes METTL3 in IECs ( Mettl3 ΔIEC) were resistant to RV infection and showed increased expression of interferons (IFNs) and IFN-stimulated genes (ISGs). Using RNA-sequencing and m6A RNA immuno-precipitation (RIP)-sequencing, we identified IRF7, a master regulator of IFN responses, as a primary target of m6A during virus infection. In the absence of METTL3, IECs showed increased Irf7 mRNA stability and enhanced type I and III IFN expression. Deficiency in IRF7 attenuated the elevated expression of IFNs and ISGs and restored susceptibility to RV infection in Mettl3 ΔIEC mice. Moreover, the global frequency of m6A modifications on mRNA transcripts declined with age in mice, with a significant drop during the period from 2 weeks to 3 weeks after birth, which is likely to have broad implications for the development of the intestinal immune system against enteric viruses early in life. Collectively, our results demonstrate a novel host m6A-IRF7-IFN antiviral signaling cascade that restricts rotavirus infection in vivo.
Article activity feed
-

-

Author Response:
Reviewer #1 (Public Review):
Wang et al., investigated the role of RNA m6A modification in intestinal epithelial cells (IECs) in the context of rotavirus infection. The authors found that the mice which specifically lacks METTL3 in IECs show resistance to rotavirus infection. They attributed this effect to increased IFN and ISG expression presumably via IRF7 upregulation. Further genetic IRF7 ablation in IECs led to the sensitivity rotavirus infection. They also found that ALKBH5 is suppressed by a rotaviral protein, although the knockout of ALKBH5 in IECs did not influence viral infection.
Overall, although the resistance of IEC-specific METTL3-deficient mice upon rotavirus infection via the control of IRF7 is a novel and interesting finding, the proposed model is not fully supported by the findings here. …
Author Response:
Reviewer #1 (Public Review):
Wang et al., investigated the role of RNA m6A modification in intestinal epithelial cells (IECs) in the context of rotavirus infection. The authors found that the mice which specifically lacks METTL3 in IECs show resistance to rotavirus infection. They attributed this effect to increased IFN and ISG expression presumably via IRF7 upregulation. Further genetic IRF7 ablation in IECs led to the sensitivity rotavirus infection. They also found that ALKBH5 is suppressed by a rotaviral protein, although the knockout of ALKBH5 in IECs did not influence viral infection.
Overall, although the resistance of IEC-specific METTL3-deficient mice upon rotavirus infection via the control of IRF7 is a novel and interesting finding, the proposed model is not fully supported by the findings here. Especially, the following points need to be addressed:
We are grateful to the reviewer for the complimentary summary of our research. We also appreciate the valuable experiments suggested by the reviewer to improve our manuscript. We have added additional important controls and mechanistic data to further support our conclusions.
- The m6A dot blot used in Figure 1 is not a good measurement system of total m6A modification levels, because the antibody used here also detects other RNA modification, m6Am (PMID: 31676230). Therefore, it is unclear if the increase of m6A dot blot intensity is due to the increase of m6A in RNAs mediated by METTL3 in IECs. The authors should investigate the m6A levels in IECs, not BMDMs, under METTL3 deficiency. Ideally, this analysis should be done using mass spectrometry.
We thank the reviewer for raising a critical point. We have tried several methods to avoid the potential non-specific detection of the previous antibody (Synaptic System, #202003) we used, which was reported to detect m6Am as well.
1.We have included Dot Blot data for m6A modification in Mettl3^△IEC and WT IECs during RV infection by using another m6A antibody (Anti-N6-methyladenosine (m6A), Sigma-Aldrich, Cat. No. ABE572-I). (see below and also Fig. 1d, 1e)
2.We have included mass spectrometry data for m6A modification in IECs during development (see below and also Fig. 1c) or RV infection (see below and also Fig. s3a).
These data suggested m6A modifications in IECs are indeed regulated during the development or RV infection. We have included the descriptions in the text.
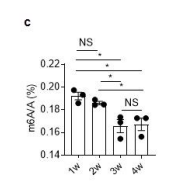
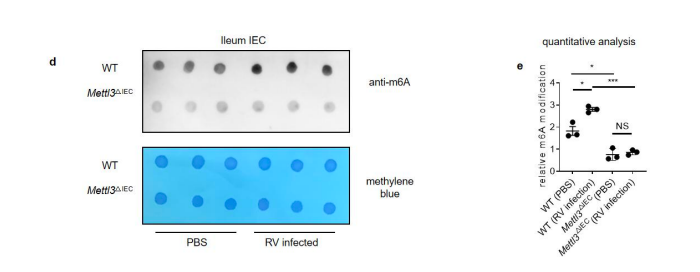
Figure 1. Rotavirus infection increases global m6A modifications, and Mettl3 deficiency in intestinal epithelial cells results in increased resistance to rotavirus infection. (c) MS analysis of m6A level in ileum tissue from mice with different ages. (mean ± SEM), Statistical significance was determined by Student’s t-test (*P < 0.05, NS., not significant). (d) WT and Mettl3^△IEC mice were infected by rotavirus EW strain at 8 days post birth. m6A dot blot analysis of total RNA in ileum IEC at 2 dpi. Methylene blue (MB) staining was the loading control. (e) Quantitative analysis of (d) (mean ± SEM). Statistical significance was determined by Student’s t-test (*P < 0.05, ***P<0.001, NS., not significant). The quantitative m6A signals were normalized to quantitative MB staining signals.
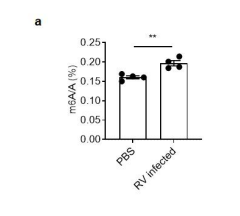
Figure s3. MS analysis of total m6A level in mice ileum. (a) WT and Mettl3 △IEC mice were infected by rotavirus EW strain at 8 days post birth. MS analysis of m6A level in ileum tissue from mice at 2 dpi (mean ± SEM), Statistical significance was determined by Student’s t-test (**P < 0.005)
- The authors show that Alkbh5 expression is increased when the mice grow up to 3 weeks old. However, the Alkbh5 protein expression changes are missing.
We thank the reviewer for raising this point. We have included the protein expression of ALKBH5 in intestine during the development (see below and Fig. s1). The ALKBH5 protein levels are increased in the intestine along with the age (Fig. s1a, s1b), which is consistent to the changes of mRNA levels of ALKBH5 during the development (Fig. 1d).
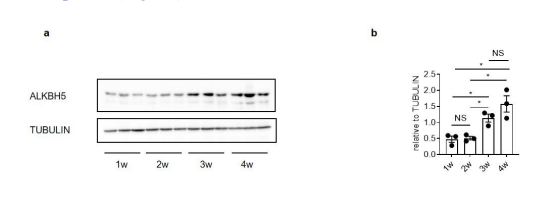
Figure s1. ALKBH5 regulate total m6A level in intestine. (a) Immunoblotting with antibodies target ALKBH5 and TUBULIN in ileum tissues from mice with different ages. (b) Quantitative analysis of (a) (mean ± SEM), Statistical significance was determined by Student’s t-test (*P < 0.05, NS., not significant).
- The authors claim that m6A declined from 2 to 2 weeks post birth is caused by increased Alkbh5 (Line 110). However, it is not clear if the subtle increase in Alkbh5 mRNA leads to the change in global m6A levels. The author can use ALKBH5-deficient mouse cells to confirm this point.
We thank the reviewer for pointing out an important point. We have included the ALKBH5 over-expression or knock-down data in a mouse IEC cell line MODE-K, to test whether the regulation of Alkbh5 mRNA in IECs leads to the change in global m6A levels.
Over-expression of ALKBH5 in MODE-K cells largely reduced the global m6A level (see below and Fig. s1d). 1. Crispr-mediated knock down of ALKBH5 in MODE-K cells augmented the global m6A level while knock down of another m6A eraser FTO in MODE-K cells didn’t affect the global m6A level (see below and Fig. s10b).

Figure s1. ALKBH5 regulate total m6A level in intestine. (d) Immunoblotting with antibodies target ALKBH5 and TUBULIN in MODE-K cells transfected with pSIN-EV or pSIN-mAlkbh5-3xFlag for 24h. m6A dot blot analysis of total RNA in indicated samples. Methylene blue (MB) staining was the loading control.
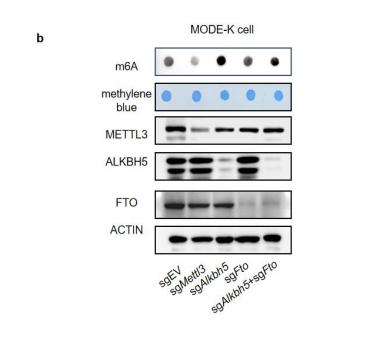
Figure s10. Alkbh5 is the dominant m6A eraser in intestine. (b) m6A dot blot analysis of total RNA in different MODE-K cells. Methylene blue (MB) staining was the loading control.
- The authors should describe the overall phenotype of IEC-specific METTL3-deficient mice at the steady state. It is important to clarify if the augmented expression of ISG upon METTL3 deficiency is dependent on rotavirus infection. Also, the authors should describe any detectable abnormalities or changes without stimulation.
We actually collaborated another group and found there is a defect in intestinal stem cells in IEC-specific METTL3-deficient mice. However, as RV normally infected IECs in the villi but not in the crypt, and stem cells are not the major producers of IFN/ISGs (Sue E. Crawford et al. Nature reviews disease primers, 2017). The defect in intestinal stem cells will less likely affect the RV infection phenotype. As it is another story that are under review, we tend to not include this part of the data in our manuscript. Moreover, we have crossed Irf7^−/− mice to Mettl3^ΔIEC mice and verified Irf7 mediated induction of ISGs is critical for the anti-viral phenotype in Mettl3^ΔIEC mice.
Our bulk RNA-seq data in IECs showed the augmented expression of ISGs upon METTL3 deficiency in steady state (Fig. 2a). We also found an augmented ISG expression in intestine of METTL3-deficient mice in steady state or early infection of RV (2d) by qPCR. However, as the RV loads in METTL3-deficient mice during the late infection stage are significantly lower than WT mice, thus the inducible ISGs expressions are consequently lower in intestine of METTL3-deficient mice than WT mice in day 4 post infection (Fig. 3f).
- The finding that IRF7 is targeted by METTL3 is not convincing. First, the authors performed MeRIP-seq and -qPCR experiments only using RNAs from wild-type IECs not from METTL3-deficient cells. It is necessary to show that the modification levels on IRF7 mRNA is indeed reduced upon METTL3 deficiency. Second, it is unclear if MeRIP-seq is properly performed or not, because there is no quality checking figure shown. For instance, the authors can generate metagene plots or gene logos of m6A modified sites to see if there is any consistency with previous reports. Third, in Figure 2h, the authors should show that the change in luciferase activity between wild-type and mutant Irf7-3'UTR reporters is dependent on METTL3 activity by performing METTL3 knockdown or knockout. Also, the authors should describe how they mutagenize the sequences for clarification. Fourth, in Figures 2F and 3C, they showed that IRF7 is upregulated in METTL3-deficient IECs while in Figure 3F, IRF7 is conversely downregulated in METTL3-deficient IECs. This is apparently contradictory to each other.
We appreciate the valuable suggestion provided by the reviewer to improve our manuscript.
- We have done RIP-qPCR in Mettl3 knock-down and WT MODE-K cells to verify the m6A modification on IRF7 mRNA, the modification levels on IRF7 mRNA is indeed reduced upon METTL3 deficiency (see below and Fig. s5c, s5d). We have added the description of the experiment in the manuscript.
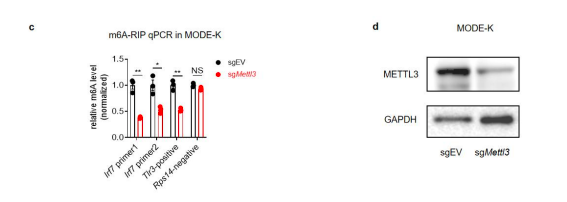
Figure s5. Characterization of m6A modifications on Irf7 mRNA. (c) m6A-RIP-qPCR confirms Irf7 as an m6A-modified gene in IECs. Fragmented RNA of sgEV and sgMettl3 MODE-K cells was incubated with an anti-m6A antibody (Sigma Aldrich ABE572-I). The eluted RNA and input were processed as described in ‘RT-qPCR’section, the data were normalized to the input samples (n=3, mean ± SEM, Statistical significance was determined by Student’s t-test (*P < 0.05, **P < 0.005, NS., not significant). Tlr3 and Rps14 were measured with m6A sites specific qPCR primer as positive control and negative control, Irf7 was measured with predicted m6A sites specific qPCR primers. (d) Knock down efficiency of METTL3 in MODE-K cells.
- We have performed metagene plots as suggested. As shown in figure s5b, the m6A peak is enriched near the stop codon and 3’UTR region, which is consistent with previously study (Xuan et al. 2018; Dominissini et al., 2012; Yang et al., 2019). We have added the description in the manuscript.
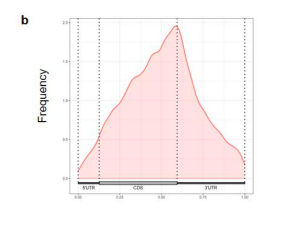
Figure s5. Characterization of m6A modfications on Irf7 mRNA. (b) Metagene plots of m6A modified sites.
- We have performed the luciferase assay in WT and METTL3 knockdown 293t cell, and found increased luciferase activity in mutant Irf7-3'UTR reporters is dependent on METTL3 activity (see below and fig. 2h, s5e). We have added the description of the experiment into the manuscript.
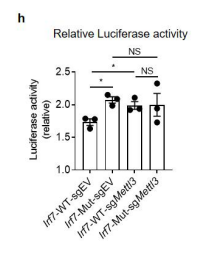
Figure 2. Mettl3 deficiency in intestinal epithelial cells results in decreased m6A deposition on Irf7, and increased interferon responses. (h) Relative luciferase activity of sgEV and sgMettl3 HEK293T cells transfected with pmirGLO-Irf7-3’UTR (Irf7-WT) or pmirGLO-Irf7-3’UTR containing mutated m6A modification sites (Irf7-MUT). The firefly luciferase activity was normalized to Renilla luciferase activity (n=3, mean ± SEM). Statistical significance was determined by Student’s t-tests between genotypes (*P < 0.05, NS., not significant).
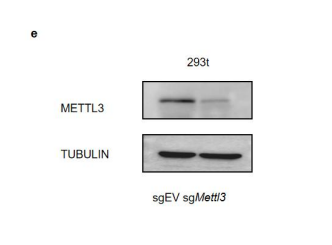
Figure s5. Characterization of m6A modifications on Irf7 mRNA. (e) Knock down efficiency of METTL3 in 293t cells used for luciferase assay.
- IRF7 is an ISG. The expression of IRF7 is controlled by both PAMP (such as virus component)-induced transcription and post-transcriptional regulation like m6A modification mediated mRNA decay. In steady state or early stage (2d) of rotavirus infection, there is no virus or the viral loads is comparable in both Mettl3^△IEC mice and WT mice, thus, IRF7 expression is mainly regulated by m6A and is higher in IECs from Mettl3^△IEC mice in comparison with that from WT mice. However, as the RV loads in Mettl3^△IEC mice during the late infection stage are significantly lower than WT mice, in this case, IRF7 expression is mainly regulated by the PAMP from virus, thus the inducible IRF7 expressions is consequently lower in intestine of Mettl3^△IEC than WT mice in day 4 post infection (Fig. 3f).
- It is unclear if the augmented expression of IRF7 per se upregulates IFN and ISG expression. Since IRF7 exerts its transcriptional activity upon phosphorylation, the authors should examine IRF7 phosphorylation and total protein levels in METTL3-deficient IECs. Also, it is interesting to see if the phosphorylation of TBK1 is augmented or not.
We have provided the phosphorylation and total protein levels of IRF7 and TBK1 in MODE-K cells treated with poly I:C. Both total IRF7 and phosphorylated IRF7 are upregulated in Mettl3-knock down cells compare to control cells (see below and Fig s5f). However, Both total TBK1 and phosphorylated TBK1 remain unchanged (Fig s5f), suggesting the augmented ISGs are less likely due to the activation of the upstream signal of IFN.
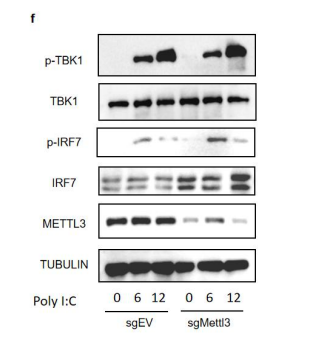
Figure s5. Characterization of m6A modifications on Irf7 mRNA. (f) Western blot analysis of sgEV and sgMettl3 MODE-K cells transfected by lipo3000 with 2ug/ml poly I:C at indicated hours post transfection, at least three replicate experiments were performed.
- In Figure 3, the authors utilized METTL3 and IRF7 deficient mice to show the contribution of METTL3-mediated IRF7 regulation in rotavirus infection. However, if IRF7 is totally abrogated, IFN production should be greatly impaired as shown in Figure 3A. Thus, it is not surprising to see that the IFN response is diminished. The authors can use heterozygous IRF7 deficient mice instead to check if upregulation of IRF7 under METTL3 deficiency is critical to control rotavirus infection.
We thank the reviewer for pointing out an important issue. However, we checked the IRF7 expression levels in IECs from Irf7^+/+ , Irf7^+/- and Irf7^-/- mice and found that there is no difference between IRF7 levels in IECs from Irf7^+/- mice and that in IECs from Irf7^+/+ mice. Thus, it is not feasible to use heterozygous IRF7 deficient mice to test the idea (Supporting Figure 1).
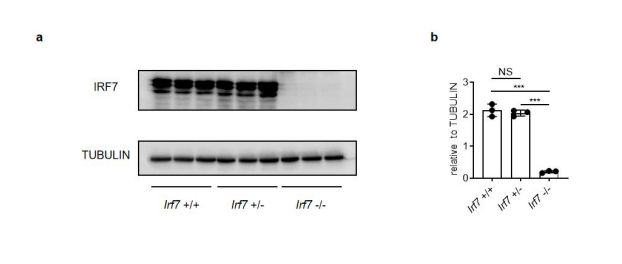
Supporting Figure 1. WT and Irf7 Heterozygous mice show same IRF7 expression level in IECs. (a) IECs from 2-weeks-old Irf7^+/+ , Irf7^+/-, Irf7^-/- mice were isolated. Western blot analysis show IRF7 expression level in different mice. (b) Quantitative analysis of (a) (mean ± SEM), statistical significance was determined by Student’s t-test ( ***P < 0.001, NS., not significant).
- Given no effect of ALKBH5 knockout on rotavirus infection as shown in Figure 4, it is questionable if ALKBH5 has a profound role in the regulation of m6A in IECs. The authors should determine if m6A modification levels are increased in IECs under ALKBH5 deficiency.
We performed the m6A dot blot assay to detect m6A modification levels in ALKBH5-knock down MODE-K cells and we do find an increase of m6A modification level under ALKBH5 deficiency (see above and Fig s10). No effect of ALKBH5 knockout on rotavirus infection actually puzzled us as well before (Fig.4c, 4d and 4e), until we found RV infection down-regulated ALKBH5 expression in the intestine of WT mice (Fig.4a).
-
Evaluation Summary:
This paper is of interest within the field of virus infection and RNA methylation. The data in the manuscript provide novel information on RNA methylation during rotavirus infection but do not fully support the conclusion because of several technical issues and experimental design.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. The reviewers remained anonymous to the authors.)
-
Reviewer #1 (Public Review):
Wang et al., investigated the role of RNA m6A modification in intestinal epithelial cells (IECs) in the context of rotavirus infection. The authors found that the mice which specifically lacks METTL3 in IECs show resistance to rotavirus infection. They attributed this effect to increased IFN and ISG expression presumably via IRF7 upregulation. Further genetic IRF7 ablation in IECs led to the sensitivity rotavirus infection. They also found that ALKBH5 is suppressed by a rotaviral protein, although the knockout of ALKBH5 in IECs did not influence viral infection.
Overall, although the resistance of IEC-specific METTL3-deficient mice upon rotavirus infection via the control of IRF7 is a novel and interesting finding, the proposed model is not fully supported by the findings here. Especially, the following …
Reviewer #1 (Public Review):
Wang et al., investigated the role of RNA m6A modification in intestinal epithelial cells (IECs) in the context of rotavirus infection. The authors found that the mice which specifically lacks METTL3 in IECs show resistance to rotavirus infection. They attributed this effect to increased IFN and ISG expression presumably via IRF7 upregulation. Further genetic IRF7 ablation in IECs led to the sensitivity rotavirus infection. They also found that ALKBH5 is suppressed by a rotaviral protein, although the knockout of ALKBH5 in IECs did not influence viral infection.
Overall, although the resistance of IEC-specific METTL3-deficient mice upon rotavirus infection via the control of IRF7 is a novel and interesting finding, the proposed model is not fully supported by the findings here. Especially, the following points need to be addressed:
The m6A dot blot used in Figure 1 is not a good measurement system of total m6A modification levels, because the antibody used here also detects other RNA modification, m6Am (PMID: 31676230). Therefore, it is unclear if the increase of m6A dot blot intensity is due to the increase of m6A in RNAs mediated by METTL3 in IECs. The authors should investigate the m6A levels in IECs, not BMDMs, under METTL3 deficiency. Ideally, this analysis should be done using mass spectrometry.
The authors show that Alkbh5 expression is increased when the mice grow up to 3 weeks old. However, the Alkbh5 protein expression changes are missing.
The authors claim that m6A declined from 2 to 2 weeks post birth is caused by increased Alkbh5 (Line 110). However, it is not clear if the subtle increase in Alkbh5 mRNA leads to the change in global m6A levels. The author can use ALKBH5-deficient mouse cells to confirm this point.
The authors should describe the overall phenotype of IEC-specific METTL3-deficient mice at the steady state. It is important to clarify if the augmented expression of ISG upon METTL3 deficiency is dependent on rotavirus infection. Also, the authors should describe any detectable abnormalities or changes without stimulation.
The finding that IRF7 is targeted by METTL3 is not convincing. First, the authors performed MeRIP-seq and -qPCR experiments only using RNAs from wild-type IECs not from METTL3-deficient cells. It is necessary to show that the modification levels on IRF7 mRNA is indeed reduced upon METTL3 deficiency. Second, it is unclear if MeRIP-seq is properly performed or not, because there is no quality checking figure shown. For instance, the authors can generate metagene plots or gene logos of m6A modified sites to see if there is any consistency with previous reports. Third, in Figure 2h, the authors should show that the change in luciferase activity between wild-type and mutant Irf7-3'UTR reporters is dependent on METTL3 activity by performing METTL3 knockdown or knockout. Also, the authors should describe how they mutagenize the sequences for clarification. Fourth, in Figures 2F and 3C, they showed that IRF7 is upregulated in METTL3-deficient IECs while in Figure 3F, IRF7 is conversely downregulated in METTL3-deficient IECs. This is apparently contradictory to each other.
It is unclear if the augmented expression of IRF7 per se upregulates IFN and ISG expression. Since IRF7 exerts its transcriptional activity upon phosphorylation, the authors should examine IRF7 phosphorylation and total protein levels in METTL3-deficient IECs. Also, it is interesting to see if the phosphorylation of TBK1 is augmented or not.
In Figure 3, the authors utilized METTL3 and IRF7 deficient mice to show the contribution of METTL3-mediated IRF7 regulation in rotavirus infection. However, if IRF7 is totally abrogated, IFN production should be greatly impaired as shown in Figure 3A. Thus, it is not surprising to see that the IFN response is diminished. The authors can use heterozygous IRF7 deficient mice instead to check if upregulation of IRF7 under METTL3 deficiency is critical to control rotavirus infection.
Given no effect of ALKBH5 knockout on rotavirus infection as shown in Figure 4, it is questionable if ALKBH5 has a profound role in the regulation of m6A in IECs. The authors should determine if m6A modification levels are increased in IECs under ALKBH5 deficiency.
-
Reviewer #2 (Public Review):
This manuscript sets out to determine the previously uncharacterized relationship between rotavirus (RV) infection and m6A modification during intestinal infection by using a collection of both in vivo and in vitro models. The authors found m6A modification inversely correlates with resistance to RV. The increased resistance to RV is due to elevated anti-viral IFN responses in a host with diminished m6A. They investigate mechanism by RNASeq and m6A-RIP-Seq and identified IRF7 as the proposed major mediator of this anti-viral response in the m6A deficient setting. During RV infection, the virus up-regulates m6A modifications to limit the IFN response, potentially targeting m6A eraser ALKBH5 for degradation by viral NSP1 protease.
The primary finding of this manuscript is novel and intriguing, as it broadens …
Reviewer #2 (Public Review):
This manuscript sets out to determine the previously uncharacterized relationship between rotavirus (RV) infection and m6A modification during intestinal infection by using a collection of both in vivo and in vitro models. The authors found m6A modification inversely correlates with resistance to RV. The increased resistance to RV is due to elevated anti-viral IFN responses in a host with diminished m6A. They investigate mechanism by RNASeq and m6A-RIP-Seq and identified IRF7 as the proposed major mediator of this anti-viral response in the m6A deficient setting. During RV infection, the virus up-regulates m6A modifications to limit the IFN response, potentially targeting m6A eraser ALKBH5 for degradation by viral NSP1 protease.
The primary finding of this manuscript is novel and intriguing, as it broadens the role of this important type of RNA modification. However, there are concerns as to whether the mechanistic conclusions are sufficiently supported. Given the central role of IRF7 in the interferon response, it is perhaps not surprising that removing IRF7 reverses the resistance to RV displayed by mettl3 mutant mice. As the authors mention in their discussion, m6A modification of RV RNA, endogenous retroviral elements, and IFN transcripts can all contribute to the decreased anti-viral response conferred by this modification. IRF7 is an ISG itself and could mediate these other mechanisms.
-
