Nuclear SUN1 stabilizes endothelial cell junctions via microtubules to regulate blood vessel formation
Curation statements for this article:-
Curated by eLife

eLife assessment
Endothelial cells mediate the growth of the vascular system, but they also need to prevent vascular leakage, which involves interactions with neighboring endothelial cells through junctional protein complexes. This important study provides a full mechanistic insight into how Sun1 is achieving its function, which supports the concept that nuclear anchoring is critical for proper mechanosensing and junctional organization. Although the evidence supporting the claims of the authors is solid and there are several merits and strengths in this study, a weakness is that some important controls are missing. The work will be of broad interest to cell biologists and vascular biologists.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Endothelial cells line all blood vessels, where they coordinate blood vessel formation and the blood-tissue barrier via regulation of cell-cell junctions. The nucleus also regulates endothelial cell behaviors, but it is unclear how the nucleus contributes to endothelial cell activities at the cell periphery. Here, we show that the nuclear-localized li nker of the n ucleoskeleton and c ytoskeleton (LINC) complex protein SUN1 regulates vascular sprouting and endothelial cell-cell junction morphology and function. Loss of murine endothelial Sun1 impaired blood vessel formation and destabilized junctions, angiogenic sprouts formed but retracted in SUN1-depleted sprouts, and zebrafish vessels lacking Sun1b had aberrant junctions and defective cell-cell connections. At the cellular level, SUN1 stabilized endothelial cell-cell junctions, promoted junction function, and regulated contractility. Mechanistically, SUN1 depletion altered cell behaviors via the cytoskeleton without changing transcriptional profiles. Reduced peripheral microtubule density, fewer junction contacts, and increased catastrophes accompanied SUN1 loss, and microtubule depolymerization phenocopied effects on junctions. Depletion of GEF-H1, a microtubule-regulated Rho activator, or the LINC complex protein nesprin-1 rescued defective junctions of SUN1-depleted endothelial cells. Thus, endothelial SUN1 regulates peripheral cell-cell junctions from the nucleus via LINC complex-based microtubule interactions that affect peripheral microtubule dynamics and Rho-regulated contractility, and this long-range regulation is important for proper blood vessel sprouting and junction integrity.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
Buglak et al. describe a role for the nuclear envelope protein Sun1 in endothelial mechanotransduction and vascular development. The study provides a full mechanistic investigation of how Sun1 is achieving its function, which supports the concept that nuclear anchoring is important for proper mechanosensing and junctional organization. The experiments have been well designed and were quantified based on independent experiments. The experiments are convincing and of high quality and include Sun1 depletion in endothelial cell cultures, zebrafish, and in endothelial-specific inducible knockouts in mice.
We thank the reviewer for their enthusiastic comments and for noting our use of multiple model systems.
Reviewer #2 (Public Review):
Endothelial cells mediate the growth of the vascular …
Author Response
Reviewer #1 (Public Review):
Buglak et al. describe a role for the nuclear envelope protein Sun1 in endothelial mechanotransduction and vascular development. The study provides a full mechanistic investigation of how Sun1 is achieving its function, which supports the concept that nuclear anchoring is important for proper mechanosensing and junctional organization. The experiments have been well designed and were quantified based on independent experiments. The experiments are convincing and of high quality and include Sun1 depletion in endothelial cell cultures, zebrafish, and in endothelial-specific inducible knockouts in mice.
We thank the reviewer for their enthusiastic comments and for noting our use of multiple model systems.
Reviewer #2 (Public Review):
Endothelial cells mediate the growth of the vascular system but they also need to prevent vascular leakage, which involves interactions with neighboring endothelial cells (ECs) through junctional protein complexes. Buglak et al. report that the EC nucleus controls the function of cell-cell junctions through the nuclear envelope-associated proteins SUN1 and Nesprin-1. They argue that SUN1 controls microtubule dynamics and junctional stability through the RhoA activator GEF-H1.
In my view, this study is interesting and addresses an important but very little-studied question, namely the link between the EC nucleus and cell junctions in the periphery. The study has also made use of different model systems, i.e. genetically modified mice, zebrafish, and cultured endothelial cells, which confirms certain findings and utilizes the specific advantages of each model system. A weakness is that some important controls are missing. In addition, the evidence for the proposed molecular mechanism should be strengthened.
We thank the reviewer for their interest in our work and for highlighting the relative lack of information regarding connections between the EC nucleus and cell periphery, and for noting our use of multiple model systems. We thank the reviewer for suggesting additional controls and mechanistic support, and we have made the revisions described below.
Specific comments:
- Data showing the efficiency of Sun1 inactivation in the murine endothelial cells is lacking. It would be best to see what is happening on the protein level, but it would already help a great deal if the authors could show a reduction of the transcript in sorted ECs. The excision of a DNA fragment shown in the lung (Fig. 1-suppl. 1C) is not quantitative at all. In addition, the gel has been run way too short so it is impossible to even estimate the size of the DNA fragment.
We agree that the DNA excision is not sufficient to demonstrate excision efficiency. We attempted examination of SUN1 protein levels in mutant retinas via immunofluorescence, but to date we have not found a SUN1 antibody that works in mouse retinal explants. We argue that mouse EC isolation protocols enrich but don’t give 100% purity, so that RNA analysis of lung tissue also has caveats. Finally, we contend that our demonstration of a consistent vascular phenotype in Sun1iECKO mutant retinas argues that excision has occurred. To test the efficiency of our excision protocol, we bred Cdh5CreERT2 mice with the ROSAmT/mG excision reporter (cells express tdTomato absent Cre activity and express GFP upon Cre-mediated excision (Muzumdar et al., 2007). Utilizing the same excision protocol as used for the Sun1iECKO mice, we see a significantly high level of excision in retinal vessels only in the presence of Cdh5CreERT2 (Reviewer Figure 1).
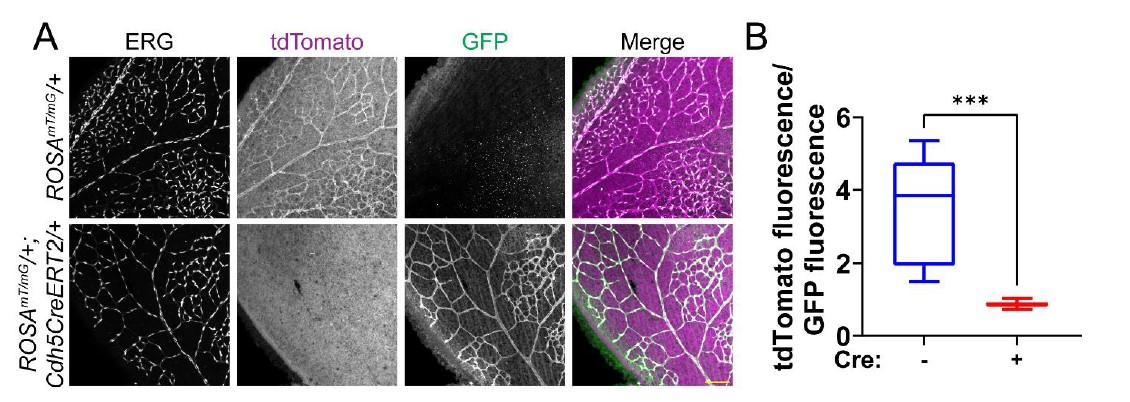
Reviewer Figure 1: Cdh5CreERT2 efficiently excises in endothelial cells of the mouse postnatal retina. (A) Representative images of P7 mouse retinas with the indicated genotypes, stained for ERG (white, nucleus). tdTomato (magenta) is expressed in cells that have not undergone Cre-mediated excision, while GFP (green) is expressed in excised cells. Scale bar, 100μm. (B) Quantification of tdTomato fluorescence relative to GFP fluorescence as shown in A. tdTomato and GFP fluorescence of endothelial cells was measured by creating a mask of the ERG channel. n=3 mice per genotype. ***, p<0.001 by student’s two-tailed unpaired t-test.
- The authors show an increase in vessel density in the periphery of the growing Sun1 mutant retinal vasculature. It would be important to add staining with a marker labelling EC nuclei (e.g. Erg) because higher vessel density might reflect changes in cell size/shape or number, which has also implications for the appearance of cell-cell junctions. More ECs crowded within a small area are likely to have more complicated junctions. Furthermore, it would be useful and straightforward to assess EC proliferation, which is mentioned later in the experiments with cultured ECs but has not been addressed in the in vivo part.
We concur that ERG staining is important to show any changes in nuclear shape or cell density in the post-natal retina. We now include this data in Figure1-figure supplement 1F-G. We do not see obvious changes in nuclear shape or number, though we do observe some crowding in Sun1iECKO retinas, consistent with increased density. However, when normalized to total vessel area, we do not observe a significant difference in the nuclear signal density in Sun1iECKO mutant retinas relative to controls.
- It appears that the loss of Sun1/sun1b in mice and zebrafish is compatible with major aspects of vascular growth and leads to changes in filopodia dynamics and vascular permeability (during development) without severe and lasting disruption of the EC network. It would be helpful to know whether the loss-of-function mutants can ultimately form a normal vascular network in the retina and trunk, respectively. It might be sufficient to mention this in the text.
We thank the reviewer for pointing this out. It is true that developmental defects in the vasculature resulting from various genetic mutations are often resolved over time. We’ve made text changes to discuss viability of Sun1 global KO mice and lack of perduring effects in sun1 morphant fish, perhaps resulting from compensation by SUN2, which is partially functionally redundant with SUN1 in vivo (Lei et al., 2009; Zhang, et al., 2009) (p. 20).
- The only readout after the rescue of the SUN1 knockdown by GEF-H1 depletion is the appearance of VE-cadherin+ junctions (Fig. 6G and H). This is insufficient evidence for a relatively strong conclusion. The authors should at least look at microtubules. They might also want to consider the activation status of RhoA as a good biochemical readout. It is argued that RhoA activity goes up (see Fig. 7C) but there is no data supporting this conclusion. It is also not clear whether "diffuse" GEF-H1 localization translates into increased Rho A activity, as is suggested by the Rho kinase inhibition experiment. GEF-H1 levels in the Western blot in (Fig. 6- supplement 2C) have not been quantitated.
We agree that analysis of RhoA activity and additional analysis of rescued junctions strengthens our conclusions, so we performed these experiments. New data (Figure 6IJ) shows that co-depletion of SUN1 and GEF-H1 rescues junction integrity as measured by biotin-matrix labeling. Interestingly, co-depletion of SUN1 and GEF-H1 does not rescue reduced microtubule density at the periphery (Figure 6-figure supplement 3BC), placing GEF-H1 downstream of aberrant microtubule dynamics in SUN1 depleted cells. This is consistent with our model (Figure 8) describing how loss of SUN1 leads to increased microtubule depolymerization, resulting in release and activation of GEF-H1 that goes on to affect actomyosin contractility and junction integrity. In addition, we include images of the junctions in GEF-H1 single KD (Figure 6-figure supplement 3BC) and quantify the western blot in Figure 6-figure supplement 3A.
We performed RhoA activity assays and new data shows that SUN1 depletion results in increased RhoA activation, while co-depletion of SUN1 and GEF-H1 ameliorates this increase (Figure 6-figure supplement 2D). This is consistent with our model in which loss of SUN1 leads to increased RhoA activity via release of GEF-H1 from microtubules. In addition, we now cite a recent study describing that GEF-H1 is activated when unbound to microtubules, with this activation resulting in increased RhoA activity (Azoitei et al., 2019).
- The criticism raised for the GEF-H1 rescue also applies to the co-depletion of SUN1 and Nesprin-1. This mechanistic aspect is currently somewhat weak and should be strengthened. Again, Rho A activity might be a useful and quantitative biochemical readout.
We respectfully point out that we showed that co-depletion of nesprin-1 and SUN1 rescues SUN1 knockdown effects via several readouts, including rescue of junction morphology, biotin labeling, microtubule localization at the periphery, and GEFH1/microtubule localization. We’ve moved this data to the main figure (Figure 7B-C, E-F) to better highlight these mechanistic findings. These results are consistent with our model that nesprin-1 effects are upstream of GEF-H1 localization. We also added results showing that nesprin-1 knockdown alone does not affect junction integrity, microtubule density, or GEF-H1/microtubule localization (Figure 7-figure supplement 1B-G).
Reviewer #3 (Public Review):
Here, Buglak and coauthors describe the effect of Sun1 deficiency on endothelial junctions. Sun1 is a component of the LINC complex, connecting the inner nuclear membrane with the cytoskeleton. The authors show that in the absence of Sun1, the morphology of the endothelial adherens junction protein VE-cadherin is altered, indicative of increased internalization of VE-cadherin. The change in VE-cadherin dynamics correlates with decreased angiogenic sprouting as shown using in vivo and in vitro models. The study would benefit from a stricter presentation of the data and needs additional controls in certain analyses.
We thank the reviewer for their insightful comments, and in response we have performed the revisions described below.
- The authors implicate the changes in VE-cadherin morphology to be of consequence for "barrier function" and mention barrier function frequently throughout the text, for example in the heading on page 12: "SUN1 stabilizes endothelial cell-cell junctions and regulates barrier function". The concept of "barrier" implies the ability of endothelial cells to restrict the passage of molecules and cells across the vessel wall. This is tested only marginally (Suppl Fig 1F) and these data are not quantified. Increased leakage of 10kDa dextran in a P6-7 Sun1-deficient retina as shown here probably reflects the increased immaturity of the Sun1-deficient retinal vasculature. From these data, the authors cannot state that Sun1 regulates the barrier or barrier function (unclear what exactly the authors refer to when they make a distinction between the barrier as such on the one hand and barrier function on the other). The authors can, if they do more experiments, state that loss of Sun1 leads to increased leakage in the early postnatal stages in the retina. However, if they wish to characterize the vascular barrier, there is a wide range of other tissue that should be tested, in the presence and absence of disease. Moreover, a regulatory role for Sun1 would imply that Sun1 normally, possibly through changes in its expression levels, would modulate the barrier properties to allow more or less leakage in different circumstances. However, no such data are shown. The authors would need to go through their paper and remove statements regarding the regulation of the barrier and barrier function since these are conclusions that lack foundation.
We thank the reviewer for pointing out that the language used regarding the function and integrity of the junctions is confusing, although we suggest that the endothelial cell properties measured by our assays are typically equated with “barrier function” in the literature. However, we have edited our language to precisely describe our results as suggested by the reviewer.
- In Fig 6g, the authors show that "depletion of GEF-H1 in endothelial cells that were also depleted for SUN1 rescued the destabilized cell-cell junctions observed with SUN1 KD alone". However, it is quite clear that Sun1 depletion also affects cell shape and cell alignment and this is not rescued by GEF-H1 depletion (Fig 6g). This should be described and commented on. Moreover please show the effects of GEF-H1 alone.
We thank the reviewer for pointing out the effects on cell shape. SUN1 depletion typically leads to shape changes consistent with elevated contractility, but this is considered to be downstream of the effects quantified here. We updated the panel in Figure 6G to a more representative image showing cell shape rescue by co-depletion of SUN1 and GEF-H1. We present new data panels showing that GEF-H1 depletion alone does not affect junction integrity (Figure 6I-J). We also present new data showing that co-depletion of GEF-H1 and SUN1 does not rescue microtubule density at the periphery (Figure 6-figure supplement 3B-C), consistent with our model that GEF-H1 activation is downstream of microtubule perturbations induced by SUN1 loss.
- In Fig. 6a, the authors show rescue of junction morphology in Sun1-depleted cells by deletion of Nesprin1. The effect of Nesprin1 KD alone is missing.
We thank the reviewer for this comment, and we now include new panels (Figure 7figure supplement 1B-G) demonstrating that Nesprin-1 depletion does not affect biotin-matrix labeling, peripheral microtubule density, or GEF-H1/microtubule localization absent co-depletion with SUN1. These findings are consistent with our model that Nesprin-1 loss does not affect cell junctions on its own because it is held in a non-functional complex with SUN1 that is not available in the absence of SUN1.
References
Azoitei, M. L., Noh, J., Marston, D. J., Roudot, P., Marshall, C. B., Daugird, T. A., Lisanza, S. L., Sandί, M., Ikura, M., Sondek, J., Rottapel, R., Hahn, K. M., Danuser, & Danuser, G. (2019). Spatiotemporal dynamics of GEF-H1 activation controlled by microtubule- and Src-mediated pathways. Journal of Cell Biology, 218(9), 3077-3097. https://doi.org/10.1083/jcb.201812073
Denis, K. B., Cabe, J. I., Danielsson, B. E., Tieu, K. V, Mayer, C. R., & Conway, D. E. (2021). The LINC complex is required for endothelial cell adhesion and adaptation to shear stress and cyclic stretch. Molecular Biology of the Cell, mbcE20110698. https://doi.org/10.1091/mbc.E20-11-0698
King, S. J., Nowak, K., Suryavanshi, N., Holt, I., Shanahan, C. M., & Ridley, A. J. (2014). Nesprin-1 and nesprin-2 regulate endothelial cell shape and migration. Cytoskeleton (Hoboken, N.J.), 71(7), 423–434. https://doi.org/10.1002/cm.21182
Lei, K., Zhang, X., Ding, X., Guo, X., Chen, M., Zhu, B., Xu, T., Zhuang, Y., Xu, R., & Han, M. (2009). SUN1 and SUN2 play critical but partially redundant roles in anchoring nuclei in skeletal muscle cells in mice. PNAS, 106(25), 10207–10212.
Muzumdar, M. D., Tasic, B., Miyamichi, K., Li, L., & Luo, L. (2007). A global doublefluorescent Cre reporter mouse. Genesis, 45(9), 593-605. https://doi.org/10.1002/dvg.20335
Ueda, N., Maekawa, M., Matsui, T. S., Deguchi, S., Takata, T., Katahira, J., Higashiyama, S., & Hieda, M. (2022). Inner Nuclear Membrane Protein, SUN1, is Required for Cytoskeletal Force Generation and Focal Adhesion Maturation. Frontiers in Cell and Developmental Biology, 10, 885859. https://doi.org/10.3389/fcell.2022.885859
Zhang, X., Lei, K., Yuan, X., Wu, X., Zhuang, Y., Xu, T., Xu, R., & Han, M. (2009). SUN1/2 and Syne/Nesprin-1/2 complexes connect centrosome to the nucleus during neurogenesis and neuronal migration in mice. Neuron, 64(2), 173–187. https://doi.org/10.1016/j.neuron.2009.08.018.
-
eLife assessment
Endothelial cells mediate the growth of the vascular system, but they also need to prevent vascular leakage, which involves interactions with neighboring endothelial cells through junctional protein complexes. This important study provides a full mechanistic insight into how Sun1 is achieving its function, which supports the concept that nuclear anchoring is critical for proper mechanosensing and junctional organization. Although the evidence supporting the claims of the authors is solid and there are several merits and strengths in this study, a weakness is that some important controls are missing. The work will be of broad interest to cell biologists and vascular biologists.
-
Reviewer #1 (Public Review):
Buglak et al. describe a role for the nuclear envelope protein Sun1 in endothelial mechanotransduction and vascular development. The study provides a full mechanistic investigation of how Sun1 is achieving its function, which supports the concept that nuclear anchoring is important for proper mechanosensing and junctional organization. The experiments have been well designed and were quantified based on independent experiments. The experiments are convincing and of high quality and include Sun1 depletion in endothelial cell cultures, zebrafish, and in endothelial-specific inducible knockouts in mice.
-
Reviewer #2 (Public Review):
Endothelial cells mediate the growth of the vascular system but they also need to prevent vascular leakage, which involves interactions with neighboring endothelial cells (ECs) through junctional protein complexes. Buglak et al. report that the EC nucleus controls the function of cell-cell junctions through the nuclear envelope-associated proteins SUN1 and Nesprin-1. They argue that SUN1 controls microtubule dynamics and junctional stability through the RhoA activator GEF-H1.
In my view, this study is interesting and addresses an important but very little-studied question, namely the link between the EC nucleus and cell junctions in the periphery. The study has also made use of different model systems, i.e. genetically modified mice, zebrafish, and cultured endothelial cells, which confirms certain findings …
Reviewer #2 (Public Review):
Endothelial cells mediate the growth of the vascular system but they also need to prevent vascular leakage, which involves interactions with neighboring endothelial cells (ECs) through junctional protein complexes. Buglak et al. report that the EC nucleus controls the function of cell-cell junctions through the nuclear envelope-associated proteins SUN1 and Nesprin-1. They argue that SUN1 controls microtubule dynamics and junctional stability through the RhoA activator GEF-H1.
In my view, this study is interesting and addresses an important but very little-studied question, namely the link between the EC nucleus and cell junctions in the periphery. The study has also made use of different model systems, i.e. genetically modified mice, zebrafish, and cultured endothelial cells, which confirms certain findings and utilizes the specific advantages of each model system. A weakness is that some important controls are missing. In addition, the evidence for the proposed molecular mechanism should be strengthened.
Specific comments:
Data showing the efficiency of Sun1 inactivation in the murine endothelial cells is lacking. It would be best to see what is happening on the protein level, but it would already help a great deal if the authors could show a reduction of the transcript in sorted ECs. The excision of a DNA fragment shown in the lung (Fig. 1-suppl. 1C) is not quantitative at all. In addition, the gel has been run way too short so it is impossible to even estimate the size of the DNA fragment.
The authors show an increase in vessel density in the periphery of the growing Sun1 mutant retinal vasculature. It would be important to add staining with a marker labelling EC nuclei (e.g. Erg) because higher vessel density might reflect changes in cell size/shape or number, which has also implications for the appearance of cell-cell junctions. More ECs crowded within a small area are likely to have more complicated junctions.
Furthermore, it would be useful and straightforward to assess EC proliferation, which is mentioned later in the experiments with cultured ECs but has not been addressed in the in vivo part.It appears that the loss of Sun1/sun1b in mice and zebrafish is compatible with major aspects of vascular growth and leads to changes in filopodia dynamics and vascular permeability (during development) without severe and lasting disruption of the EC network. It would be helpful to know whether the loss-of-function mutants can ultimately form a normal vascular network in the retina and trunk, respectively. It might be sufficient to mention this in the text.
The only readout after the rescue of the SUN1 knockdown by GEF-H1 depletion is the appearance of VE-cadherin+ junctions (Fig. 6G and H). This is insufficient evidence for a relatively strong conclusion. The authors should at least look at microtubules. They might also want to consider the activation status of RhoA as a good biochemical readout. It is argued that RhoA activity goes up (see Fig. 7C) but there is no data supporting this conclusion. It is also not clear whether "diffuse" GEF-H1 localization translates into increased Rho A activity, as is suggested by the Rho kinase inhibition experiment. GEF-H1 levels in the Western blot in (Fig. 6- supplement 2C) have not been quantitated.
The criticism raised for the GEF-H1 rescue also applies to the co-depletion of SUN1 and Nesprin-1. This mechanistic aspect is currently somewhat weak and should be strengthened. Again, Rho A activity might be a useful and quantitative biochemical readout.
-
Reviewer #3 (Public Review):
Here, Buglak and coauthors describe the effect of Sun1 deficiency on endothelial junctions. Sun1 is a component of the LINC complex, connecting the inner nuclear membrane with the cytoskeleton. The authors show that in the absence of Sun1, the morphology of the endothelial adherens junction protein VE-cadherin is altered, indicative of increased internalization of VE-cadherin. The change in VE-cadherin dynamics correlates with decreased angiogenic sprouting as shown using in vivo and in vitro models. The study would benefit from a stricter presentation of the data and needs additional controls in certain analyses.
1. The authors implicate the changes in VE-cadherin morphology to be of consequence for "barrier function" and mention barrier function frequently throughout the text, for example in the heading on …
Reviewer #3 (Public Review):
Here, Buglak and coauthors describe the effect of Sun1 deficiency on endothelial junctions. Sun1 is a component of the LINC complex, connecting the inner nuclear membrane with the cytoskeleton. The authors show that in the absence of Sun1, the morphology of the endothelial adherens junction protein VE-cadherin is altered, indicative of increased internalization of VE-cadherin. The change in VE-cadherin dynamics correlates with decreased angiogenic sprouting as shown using in vivo and in vitro models. The study would benefit from a stricter presentation of the data and needs additional controls in certain analyses.
1. The authors implicate the changes in VE-cadherin morphology to be of consequence for "barrier function" and mention barrier function frequently throughout the text, for example in the heading on page 12: "SUN1 stabilizes endothelial cell-cell junctions and regulates barrier function". The concept of "barrier" implies the ability of endothelial cells to restrict the passage of molecules and cells across the vessel wall. This is tested only marginally (Suppl Fig 1F) and these data are not quantified. Increased leakage of 10kDa dextran in a P6-7 Sun1-deficient retina as shown here probably reflects the increased immaturity of the Sun1-deficient retinal vasculature. From these data, the authors cannot state that Sun1 regulates the barrier or barrier function (unclear what exactly the authors refer to when they make a distinction between the barrier as such on the one hand and barrier function on the other). The authors can, if they do more experiments, state that loss of Sun1 leads to increased leakage in the early postnatal stages in the retina. However, if they wish to characterize the vascular barrier, there is a wide range of other tissue that should be tested, in the presence and absence of disease. Moreover, a regulatory role for Sun1 would imply that Sun1 normally, possibly through changes in its expression levels, would modulate the barrier properties to allow more or less leakage in different circumstances. However, no such data are shown. The authors would need to go through their paper and remove statements regarding the regulation of the barrier and barrier function since these are conclusions that lack foundation.
2. In Fig 6g, the authors show that "depletion of GEF-H1 in endothelial cells that were also depleted for SUN1 rescued the destabilized cell-cell junctions observed with SUN1 KD alone". However, it is quite clear that Sun1 depletion also affects cell shape and cell alignment and this is not rescued by GEF-H1 depletion (Fig 6g). This should be described and commented on. Moreover please show the effects of GEF-H1 alone.
3. In Fig. 6a, the authors show rescue of junction morphology in Sun1-depleted cells by deletion of Nesprin1. The effect of Nesprin1 KD alone is missing. -
