Uncovering perturbations in human hematopoiesis associated with healthy aging and myeloid malignancies at single-cell resolution
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
The present manuscript provides a valuable single-cell transcriptomic resource to understand normal hematopoiesis in humans and the age-dependent cellular and molecular alterations. It addresses very important questions in hematopoietic stem cell biology, such as the molecular changes underlying their aging and their perturbation in the context of myelodysplastic syndrome, and will be of interest to readers in the field of hematopoiesis and associated diseases, aging, and single-cell RNA sequencing.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #2 agreed to share their name with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Early hematopoiesis is a continuous process in which hematopoietic stem and progenitor cells (HSPCs) gradually differentiate toward specific lineages. Aging and myeloid malignant transformation are characterized by changes in the composition and regulation of HSPCs. In this study, we used single-cell RNA sequencing (scRNA-seq) to characterize an enriched population of human HSPCs obtained from young and elderly healthy individuals.
Based on their transcriptional profile, we identified changes in the proportions of progenitor compartments during aging, and differences in their functionality, as evidenced by gene set enrichment analysis. Trajectory inference revealed that altered gene expression dynamics accompanied cell differentiation, which could explain aging-associated changes in hematopoiesis. Next, we focused on key regulators of transcription by constructing gene regulatory networks (GRNs) and detected regulons that were specifically active in elderly individuals. Using previous findings in healthy cells as a reference, we analyzed scRNA-seq data obtained from patients with myelodysplastic syndrome (MDS) and detected specific alterations of the expression dynamics of genes involved in erythroid differentiation in all patients with MDS such as TRIB2. In addition, the comparison between transcriptional programs and GRNs regulating normal HSPCs and MDS HSPCs allowed identification of regulons that were specifically active in MDS cases such as SMAD1, HOXA6, POU2F2, and RUNX1 suggesting a role of these transcription factors (TFs) in the pathogenesis of the disease.
In summary, we demonstrate that the combination of single-cell technologies with computational analysis tools enable the study of a variety of cellular mechanisms involved in complex biological systems such as early hematopoiesis and can be used to dissect perturbed differentiation trajectories associated with perturbations such as aging and malignant transformation. Furthermore, the identification of abnormal regulatory mechanisms associated with myeloid malignancies could be exploited for personalized therapeutic approaches in individual patients.
Article activity feed
-

-

Author Response
Reviewer #2 (Public Review):
In this manuscript, the authors performed single-cell RNA sequencing (scRNA-seq) analysis on bone marrow CD34+ cells from young and old healthy donors to understand the age-dependent cellular and molecular alterations during human hematopoiesis. Using a logistic regression classifier trained on young healthy donors, they identified cell-type composition changes in old donors, including an expansion of hematopoietic stem cells (HSCs) and a reduction of committed lymphoid and myeloid lineages. They also identified cell-type-specific molecular alterations between young and old donors and age-associated changes in differentiation trajectories and gene regulatory networks (GRNs). Furthermore, by comparing the single-cell atlas of normal hematopoiesis with that of myelodysplastic syndrome (MDS), …
Author Response
Reviewer #2 (Public Review):
In this manuscript, the authors performed single-cell RNA sequencing (scRNA-seq) analysis on bone marrow CD34+ cells from young and old healthy donors to understand the age-dependent cellular and molecular alterations during human hematopoiesis. Using a logistic regression classifier trained on young healthy donors, they identified cell-type composition changes in old donors, including an expansion of hematopoietic stem cells (HSCs) and a reduction of committed lymphoid and myeloid lineages. They also identified cell-type-specific molecular alterations between young and old donors and age-associated changes in differentiation trajectories and gene regulatory networks (GRNs). Furthermore, by comparing the single-cell atlas of normal hematopoiesis with that of myelodysplastic syndrome (MDS), they characterized cellular and molecular perturbations affecting normal hematopoiesis in MDS.
The present manuscript provides a valuable single-cell transcriptomic resource to understand normal hematopoiesis in humans and the age-dependent cellular and molecular alterations. However, their main claims are not well supported by the data presented. All results were based on computational predictions, not experimentally validated.
Major points:
- The authors constructed a regularized logistic regression trained on young donors with manually annotated cell types and predicted cell type labels of cells from old and MDS samples. As the manual annotation of cell types was implicitly assumed as ground truth in this manuscript, I'm wondering whether the predicted cell types in old and MDS samples are consistent with the manual annotation. They should apply the same strategy used in young samples for manual annotation to old and MDS samples, and evaluate how accurate their classifier is.
We performed manual annotation for each MDS sample independently, and for the 3 healthy elderly donors integrated dataset. To do so, we performed unsupervised clustering with Seurat and annotated the clusters using the same set of canonical marker genes that we used for the young data. We then analyzed the correspondences between the annotated clusters and the predictions by GLMnet. Results are shown on Figure 1a. We observe that the biggest disagreements between methods occur between adjacent identities, such as HSC and LMPP, GMP and GMP with more prominent granulocytes profile, or MEP, early and late erythroid. When we explore these disagreements along the erythroid branch, we see that they particularly occur close to the border between subpopulations (Figure 1b). This is consistent with the continuous nature of the differentiation and the difficulty to establish boundaries between cell compartments. However, we observe that miss-labeling between different hematopoietic lineages is rare.
In addition, unsupervised clustering was not always able to directly separate the data in the expected subpopulations. We can see different clusters containing the same cell types (e.g. LMPP1, LMPP2), as well as individual clusters containing cells with different identities (e.g. pDC and monocyte progenitors). This is usually due to sources of variability different to cell identity present in the data Additional, supervised finetuning by local sub clustering and merging would be needed to correct for this. On the contrary, we believe that our GLMnet-based method focusses on gene expression related to identity, resulting in a classification that is better suited for our purpose.
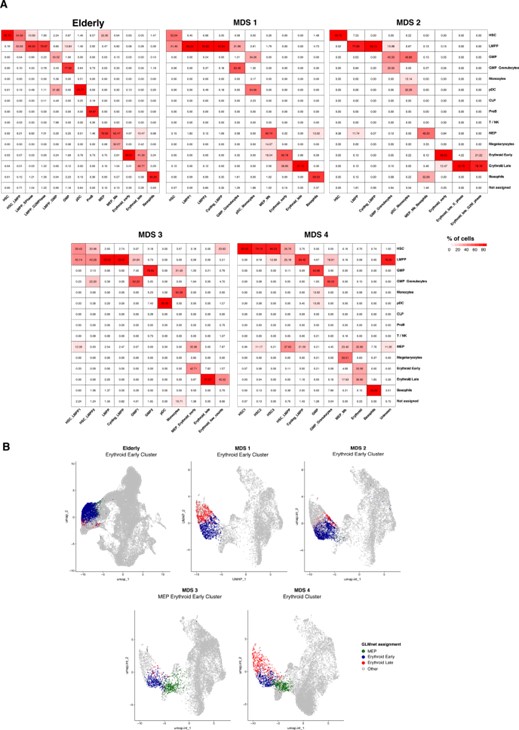
Figure 1 Comparison between GLMnet predictions and manually annotated clusters A) Heatmaps showing percentages of cells in manually annotated clusters (columns) that have been assigned to each of the cell identities predicted by our GLMnet classification method (rows). The analysis was performed independently for the elderly integrated dataset and for every MDS sample. B) UMAP plots showing disagreements in classification between adjacent cell compartments in the erythroid branch. Cells from one erythroid cluster per patient are colored by the identity assigned by the GLMnet classifier. Cells in gray are not in the highlighted cluster, nor labeled as MEP, erythroid early or erythroid late by our classifier.
- The cell-type composition changes in Figures 1 and 4 were descriptively presented without providing the statistical significance of the changes. In addition, the age-dependent cell-type composition changes should be validated by flow cytometry.
We thank the reviewer for the comment. Significance of the changes is included in Supplementary File 3. In addition, we included the percentage of several cell types we validated by flow cytometry, namely HSCs, GMPs and MEPs, in young and elderly healthy individuals in the manuscript, as Figure 1-figure supplement 3. Similarly to what we detected in our bioinformatic analyses, flow cytometry data demonstrated a significant increase in the percentage of HSCs, as well as an increasing trend in MEPs and a slight decrease in the percentage of GMPs in elderly individuals, corroborating our previous results.
- In Figure 2, the authors used two different pseudo-time inference methods, STREAM, and Palantir. It is not clear why they used two different methods for trajectory inference. Do they provide the same differentiation trajectories? How robust are the results of trajectory inference algorithms? It seems to be inconsistent that the pseudotime inferred by STREAM was not used for downstream analysis and the new pseudotime was recalculated by using Palantir.
We thank the reviewer for the comment. The reason behind using two different methods to perform similar analyses, is that each of them provides specific outputs that can be used to perform a more robust and comprehensive analysis. STREAM allows to unravel the differentiation trajectories in a single cell dataset with an unsupervised approach. Also the visualization provided by STREAM (Figure 2C and 2D) allows for a simple interpretation of the results to the reader. On the other hand, Palantir provides a more robust analysis to dissect how gene expression dynamics interact and change with differentiation trajectories. For this reason, we decided to use this second method to investigate how specific genes were altered in the monocytic compartment.
As a resource article, the showcase of different methods can be valuable as it provides examples on how each tool can be used to obtain specific results, which can help any reader to decide which might be the best tool for their specific case.
Just to confirm that pseudotime results are similar, we perform a correlation analysis with the pseudotime values obtained from each method. We observed a correlation coefficient of 0.78 (p.val < 2.2e-16) confirming the similarity among both tools.
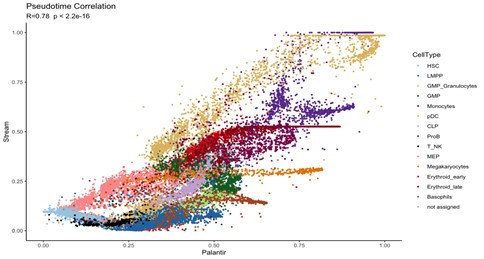
Figure 2. Correlation analysis of pseudotime values obtained with STREAM and PALANTIR.
- In Figure 2D, some HSCs seem to be committed to the erythroid lineage. The authors should carefully examine whether these HSCs are genuinely HSCS, not early erythroid progenitors.
We thank the reviewer for the comment. We have performed a deep analysis regarding the classification of HSCs (See Figure 3). Our analyses reveal that none of the cells classified as HSCs express early erythroid progenitor markers. We have also used STREAM to show the expression of these markers along the obtained trajectory and observed that erythroid markers show expression in the erythroid trajectory but not in the HSC compartment (Figure 4).
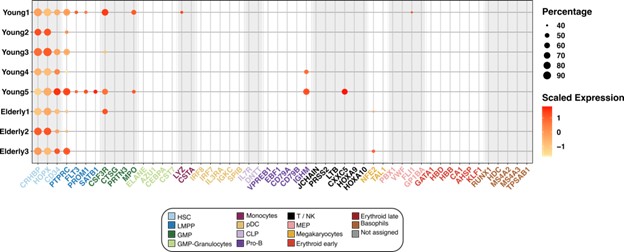
Figure 3 Expression of marker genes in the HSC compartment. Dot plot depicting the normalized scaled expression of canonical marker genes by HSC of the 5 young and 3 elderly healthy donors. Marker genes are colored by the cell population they characterize. Dot color represents expression levels, and dot size represents the percentage of cells that express a gene.
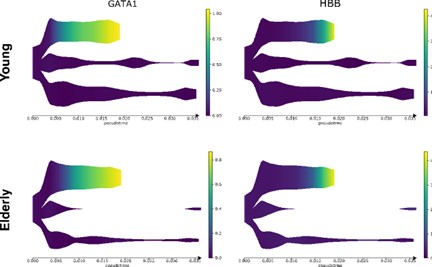
Figure 4. Expression of erythroid markers in STREAM trajectories. Expression of GATA1 and HBB (erythroid markers) in the predicted differentiation trajectories.
- It is not clear how the authors draw a conclusion from Figure 3D that the number of common targets between transcription factors is reduced. Some quantifications should be provided.
We thank the reviewer for the comment. We have updated the manuscript to better reflect our findings and emphasize that the predicted regulatory networks of HSCs in elderly donors is displayed as an independent network, compared to the young donors. (Page 6, line 36).
“Overall, we observed that the predicted regulatory network of elderly HSCs (Figure 3d) appeared as an independent network compared to the young GRN. This finding could result in the loss of co-regulatory mechanisms in the elderly donors.”
- The constructed GRNs and related descriptions were based solely on the SCENIC analysis. By providing the results of an orthogonal prediction method for GRNs, the authors should evaluate how robust and consistent their predictions are.
We thank the reviewer for the comment regarding the method to build gene regulatory networks. As a resource article, our manuscript describes a complete workflow to perform different aspects of single cell analyses. These steps go from automated classification, trajectory inference and GRN prediction. All the selected algorithms have already been benchmarked and compared against other tools that perform similar analysis. SCENIC has already been benchmarked against other algorithms (11) and by others (12).
We do agree with the reviewer that these new predictions could provide strength to our findings, however we believe that these orthogonal predictions would better fit if our article was intended for the Research Article category instead of Tools and Resources.
- The observed age-dependent cellular and molecular alterations in human hematopoiesis are interesting, but I'm wondering whether the observed alterations are driven by inflammatory microenvironment or intrinsic properties of a subpopulation of HSCs affected by clonal hematopoiesis (CH). To address this, the authors can perform genotyping of transcriptomes (GoT) on old healthy donors with CH. By comparing the transcriptomes of cells with and without CH mutations, we can evaluate the effects of CH on age-associated molecular alterations.
We thank the reviewer for the comment. Unfortunately, in order to perform GoT (genotyping of transcriptomes) on the healthy donors, requires modifying the standard 10x Genomics workflow to amplify the targeted locus and transcript of interest. This would require collecting new samples, optimizing the method and performing new analysis from scratch (from sequencing up to analysis). We believe this is not in the scope of the manuscript. On the other hand, we don’t have enough material to create new single cell libraries, this fact would require the addition of new donors and as a result, a complete new analysis to perform the integration.
Reviewer #3 (Public Review):
The authors have performed a transcriptional analysis of young/aged hematopoietic stem/progenitor cells which were obtained from normal individuals and those with MDS.
The authors generated an important and valuable dataset that will be of considerable benefit to the field. However, the data appear to be over-interpreted at times (for example, GSEA analysis does not have "functionality", as the authors claim). On the other hand, a comparison between normal-aged HSC and HSC from MDS patients appears to be under-explored in trying to understand how this disease (which is more common in the elderly) disrupts HSC function.
A more extensive cross-referencing of other normal HSPC/MDS HSCP datasets from aged humans would have been helpful to highlight the usefulness of the analytical tools that the authors have generated.
Major points
- The authors detail methodology for identification of cell types from single-cell data - GLMnet. This portion of the text needs to be clarified as it is not immediately clear what it is or how it's being used. It also needs to be explained by what metric the classifier "performed better among progenitor cell types" and why this apparent advantage was sufficient to use it for the subsequent analysis. This is critical since interpretation of the data that follows depends on the validation of GLMnet as a reliable tool.
We thank the review for the comment. We have updated the corresponding section to better describe how GLMnet is used and that the reasoning on why we decided to use GLMnet as our cell type annotation method instead of other available tools such as Seurat, is based on the results of the benchmark described in Figure 1-figure supplement 1. We also described the main differences between our method and Seurat (See Answer to Review 1, Question # 4).
- The finding of an increased number of erythroid progenitors and decreased number of myeloid cells in aged HPSC is surprising since aging is known to be associated with anemia and myeloid bias. Given that the initial validation of GLMnet is insufficiently described, this result raises concerns about the method. Along the same lines, the authors report that their tool detects a reduced frequency of monocyte progenitors. How does this finding correlate with the published data on aging humans? Is monocytopenia a feature of normal aging?
We thank the reviewer for this comment, as changes in the output of HSCs as a consequence of aging are of high interest. According to the literature, there is clear evidence of the loss of lymphoid progeny with age (13,14), which goes in agreement with our results. However, in the case of the myeloid compartment, the effects of aging are not as clear. Studies in mice have indeed observed that the loss of lymphoid cells is accompanied by increased myeloid output, starting at the level of GMPs (Rossi et al. 2005; Florian et al. 2012; Min et al. 2006). But studies on human individuals have not found changes in numbers of these myeloid progenitors (Kuranda et al. 2011; Pang et al. 2011). In addition, in the mentioned studies, myeloid production was measured exclusively by its white blood cells fraction. More recent studies have focused on the other myeloid compartments: megakaryocyte and erythroid cells. Results point towards the increase of platelet-biased HSC with age (Sanjuan-Pla et al. 2013; Grover et al. 2016) and a possible expansion of megakaryocytic and erythroid progenitor populations (Yamamoto et al. 2018; Poscablo et al. 2021; Rundberg Nilsson et al. 2016), which may represent a compensatory mechanism for the ineffective differentiation towards this lineage in elderly individuals. This goes in line with the accumulation of MEPs we see in our data. Finally, and in accordance with the reduced frequency of monocyte progenitors observed, it has been shown that with increasing age, there is a gradual decline in the monocyte count (15).
Regarding the concerns about our classification method raised by the reviewer, we have performed additional validations that we describe in answers to reviewer 1 comment #4 and reviewer 2 comment #1. To further confirm that the changes in cellular proportions we found are real, we applied two additional classification methods: Seurat transfer and Celltypist (16) to the elderly donors dataset. We obtained a similar expansion in MEPs, together with reduction of monocytic progenitors with the three methods (Figure 5).
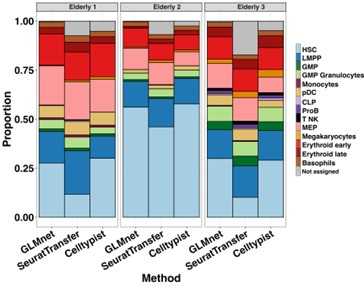
Figure 5 Classification of HSPCs from elderly donors. Barplot showing proportions of every cell subpopulation per elderly donor, resulting from three classification methods: GLMnet-based classifier, Seurat transfer and Celltypist. For the three methods, cells with prediction scores < 0,5 were labeled as “not assigned”.
- The use of terminology requires more clarity in order to better understand what kind of comparison has been performed, i.e. whether global transcriptional profiles are being compared, or those of specific subset populations. Also, the young/aged comparisons are often unclear, i.e. it's not evident whether the authors are referring to genes upregulated in aged HSC and downregulated in young HSC or vice versa. A more consistent data description would make the paper much easier to read.
We thank the reviewer for this comment. We have updated the manuscript to provide more clarity in the description of the different comparisons made in our analyses. Most changes are located in the Transcriptional profiling of human young and elderly hematopoietic progenitor systems sub-section within the Results.
- The link between aging and MDS is not explored but could be an informative use of the data that the authors have generated. For example, anemia is a feature of both aging and MDS whereas neutropenia and thrombocytopenia only occur in MDS. Are there any specific pathways governing myeloid/platelet development that are only affected in MDS?
Thank you for raising this comment. We believe that discriminating events that take place during healthy aging from those associated to MDS will be helpful to understand this particular disease, as it is so closely related to age. This is why, when analyzing MDS, we have considered young and elderly donors as two separate sets of healthy controls, the eldery donors being the most suitable one for comparisons with MDS samples.
With regards to the comment on myeloid and platelet development, the GSEA analysis gives potentially useful information. MYC targets and oxidative phosphorylation are significantly enriched in the MEP compartment from MDS patients when compared to elderly donors, indicating that these progenitors may recover a more active profile with the disease. Hypoxia related genes, on the other hand, are more active in HSCs and MEPs from healthy elderly donors than in MDS. Hypoxia is known to be implicated in megakaryocyte and erythroid differentiation (17)
- MDS is a very heterogeneous disorder and while the authors did specify that they were using samples from MDS with multilineage dysplasia, more clinical details (blood counts, cytogenetics, mutational status) are needed to be able to interpret the data.
We thank the reviewer for the comment. All the clinical details for each MDS patient are included in Supplementary File 5.
-
Evaluation Summary:
The present manuscript provides a valuable single-cell transcriptomic resource to understand normal hematopoiesis in humans and the age-dependent cellular and molecular alterations. It addresses very important questions in hematopoietic stem cell biology, such as the molecular changes underlying their aging and their perturbation in the context of myelodysplastic syndrome, and will be of interest to readers in the field of hematopoiesis and associated diseases, aging, and single-cell RNA sequencing.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #2 agreed to share their name with the authors.)
-
Reviewer #1 (Public Review):
In this study, Romero, Prosper, and colleagues have investigated the differential gene expression and regulation in hematopoietic stem and progenitor cells (HSPCs) in young or elderly healthy individuals. With the use of single-cell RNA sequencing (scRNA seq), the authors identified that the stem/progenitor repertoire is changed in elderly individuals, which is accompanied by changes in cell differentiation. The authors additionally compare HSPCs from patients with myelodysplastic syndrome (MDS) and found that MDS patients exhibit specific alterations in erythroid differentiation gene regulatory networks in MDS HSPCs. Overall, this study deals with a valuable resource of HSPC profiles in healthy individuals and proves the biased hematopoietic landscape over aging at a transcriptome level. It will serve as a …
Reviewer #1 (Public Review):
In this study, Romero, Prosper, and colleagues have investigated the differential gene expression and regulation in hematopoietic stem and progenitor cells (HSPCs) in young or elderly healthy individuals. With the use of single-cell RNA sequencing (scRNA seq), the authors identified that the stem/progenitor repertoire is changed in elderly individuals, which is accompanied by changes in cell differentiation. The authors additionally compare HSPCs from patients with myelodysplastic syndrome (MDS) and found that MDS patients exhibit specific alterations in erythroid differentiation gene regulatory networks in MDS HSPCs. Overall, this study deals with a valuable resource of HSPC profiles in healthy individuals and proves the biased hematopoietic landscape over aging at a transcriptome level. It will serve as a valuable resource for understanding the molecular basis for hematopoietic aging, which will be useful for future therapeutics and applications.
-
Reviewer #2 (Public Review):
In this manuscript, the authors performed single-cell RNA sequencing (scRNA-seq) analysis on bone marrow CD34+ cells from young and old healthy donors to understand the age-dependent cellular and molecular alterations during human hematopoiesis. Using a logistic regression classifier trained on young healthy donors, they identified cell-type composition changes in old donors, including an expansion of hematopoietic stem cells (HSCs) and a reduction of committed lymphoid and myeloid lineages. They also identified cell-type-specific molecular alterations between young and old donors and age-associated changes in differentiation trajectories and gene regulatory networks (GRNs). Furthermore, by comparing the single-cell atlas of normal hematopoiesis with that of myelodysplastic syndrome (MDS), they characterized …
Reviewer #2 (Public Review):
In this manuscript, the authors performed single-cell RNA sequencing (scRNA-seq) analysis on bone marrow CD34+ cells from young and old healthy donors to understand the age-dependent cellular and molecular alterations during human hematopoiesis. Using a logistic regression classifier trained on young healthy donors, they identified cell-type composition changes in old donors, including an expansion of hematopoietic stem cells (HSCs) and a reduction of committed lymphoid and myeloid lineages. They also identified cell-type-specific molecular alterations between young and old donors and age-associated changes in differentiation trajectories and gene regulatory networks (GRNs). Furthermore, by comparing the single-cell atlas of normal hematopoiesis with that of myelodysplastic syndrome (MDS), they characterized cellular and molecular perturbations affecting normal hematopoiesis in MDS.
The present manuscript provides a valuable single-cell transcriptomic resource to understand normal hematopoiesis in humans and the age-dependent cellular and molecular alterations. However, their main claims are not well supported by the data presented. All results were based on computational predictions, not experimentally validated.
Major points:
1. The authors constructed a regularized logistic regression trained on young donors with manually annotated cell types and predicted cell type labels of cells from old and MDS samples. As the manual annotation of cell types was implicitly assumed as ground truth in this manuscript, I'm wondering whether the predicted cell types in old and MDS samples are consistent with the manual annotation. They should apply the same strategy used in young samples for manual annotation to old and MDS samples, and evaluate how accurate their classifier is.
2. The cell-type composition changes in Figures 1 and 4 were descriptively presented without providing the statistical significance of the changes. In addition, the age-dependent cell-type composition changes should be validated by flow cytometry.
3. In Figure 2, the authors used two different pseudo-time inference methods, STREAM, and Palantir. It is not clear why they used two different methods for trajectory inference. Do they provide the same differentiation trajectories? How robust are the results of trajectory inference algorithms? It seems to be inconsistent that the pseudotime inferred by STREAM was not used for downstream analysis and the new pseudotime was recalculated by using Palantir.
4. In Figure 2D, some HSCs seem to be committed to the erythroid lineage. The authors should carefully examine whether these HSCs are genuinely HSCS, not early erythroid progenitors.
5. It is not clear how the authors draw a conclusion from Figure 3D that the number of common targets between transcription factors is reduced. Some quantifications should be provided.
6. The constructed GRNs and related descriptions were based solely on the SCENIC analysis. By providing the results of an orthogonal prediction method for GRNs, the authors should evaluate how robust and consistent their predictions are.
7. The observed age-dependent cellular and molecular alterations in human hematopoiesis are interesting, but I'm wondering whether the observed alterations are driven by inflammatory microenvironment or intrinsic properties of a subpopulation of HSCs affected by clonal hematopoiesis (CH). To address this, the authors can perform genotyping of transcriptomes (GoT) on old healthy donors with CH. By comparing the transcriptomes of cells with and without CH mutations, we can evaluate the effects of CH on age-associated molecular alterations. -
Reviewer #3 (Public Review):
The authors have performed a transcriptional analysis of young/aged hematopoietic stem/progenitor cells which were obtained from normal individuals and those with MDS.
The authors generated an important and valuable dataset that will be of considerable benefit to the field. However, the data appear to be over-interpreted at times (for example, GSEA analysis does not have "functionality", as the authors claim). On the other hand, a comparison between normal-aged HSC and HSC from MDS patients appears to be under-explored in trying to understand how this disease (which is more common in the elderly) disrupts HSC function.
A more extensive cross-referencing of other normal HSPC/MDS HSCP datasets from aged humans would have been helpful to highlight the usefulness of the analytical tools that the authors have …
Reviewer #3 (Public Review):
The authors have performed a transcriptional analysis of young/aged hematopoietic stem/progenitor cells which were obtained from normal individuals and those with MDS.
The authors generated an important and valuable dataset that will be of considerable benefit to the field. However, the data appear to be over-interpreted at times (for example, GSEA analysis does not have "functionality", as the authors claim). On the other hand, a comparison between normal-aged HSC and HSC from MDS patients appears to be under-explored in trying to understand how this disease (which is more common in the elderly) disrupts HSC function.
A more extensive cross-referencing of other normal HSPC/MDS HSCP datasets from aged humans would have been helpful to highlight the usefulness of the analytical tools that the authors have generated.
Major points
1. The authors detail methodology for identification of cell types from single-cell data - GLMnet. This portion of the text needs to be clarified as it is not immediately clear what it is or how it's being used. It also needs to be explained by what metric the classifier "performed better among progenitor cell types" and why this apparent advantage was sufficient to use it for the subsequent analysis. This is critical since interpretation of the data that follows depends on the validation of GLMnet as a reliable tool.
2. The finding of an increased number of erythroid progenitors and decreased number of myeloid cells in aged HPSC is surprising since aging is known to be associated with anemia and myeloid bias. Given that the initial validation of GLMnet is insufficiently described, this result raises concerns about the method. Along the same lines, the authors report that their tool detects a reduced frequency of monocyte progenitors. How does this finding correlate with the published data on aging humans? Is monocytopenia a feature of normal aging?
3. The use of terminology requires more clarity in order to better understand what kind of comparison has been performed, i.e. whether global transcriptional profiles are being compared, or those of specific subset populations. Also, the young/aged comparisons are often unclear, i.e. it's not evident whether the authors are referring to genes upregulated in aged HSC and downregulated in young HSC or vice versa. A more consistent data description would make the paper much easier to read.
4. The link between aging and MDS is not explored but could be an informative use of the data that the authors have generated. For example, anemia is a feature of both aging and MDS whereas neutropenia and thrombocytopenia only occur in MDS. Are there any specific pathways governing myeloid/platelet development that are only affected in MDS?
5. MDS is a very heterogeneous disorder and while the authors did specify that they were using samples from MDS with multilineage dysplasia, more clinical details (blood counts, cytogenetics, mutational status) are needed to be able to interpret the data.
-
-
-
-
-
