The oncoprotein BCL6 enables solid tumor cells to evade genotoxic stress
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
This research explored the role that BCL6 exerts on chemotherapy resistance in solid tumors other than previously reported hematological malignancies. The study also implicates the IFN-BCL6-PTEN axis as an antagonism target for overcoming resistance. This paper will be interesting to scientists who are specialists in cancer chemoresistance and oncological pharmacology.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #3 agreed to share their name with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Genotoxic agents remain the mainstay of cancer treatment. Unfortunately, the clinical benefits are often countered by a rapid tumor adaptive response. Here, we report that the oncoprotein B cell lymphoma 6 (BCL6) is a core component that confers solid tumor adaptive resistance to genotoxic stress. Multiple genotoxic agents promoted BCL6 transactivation, which was positively correlated with a weakened therapeutic efficacy and a worse clinical outcome. Mechanistically, we discovered that treatment with the genotoxic agent etoposide led to the transcriptional reprogramming of multiple pro-inflammatory cytokines, among which the interferon-α and interferon-γ responses were substantially enriched in resistant cells. Our results further revealed that the activation of interferon/signal transducer and activator of transcription 1 axis directly upregulated BCL6 expression. The increased expression of BCL6 further repressed the tumor suppressor PTEN and consequently enabled resistant cancer cell survival. Accordingly, targeted inhibition of BCL6 remarkably enhanced etoposide-triggered DNA damage and apoptosis both in vitro and in vivo. Our findings highlight the importance of BCL6 signaling in conquering solid tumor tolerance to genotoxic stress, further establishing a rationale for a combined approach with genotoxic agents and BCL6-targeted therapy.
Article activity feed
-

-

Author Response:
Reviewer #2 (Public Review):
In the paper entitled "The Oncoprotein BCL6 Enables Cancer Cells to Evade Genotoxic Stress", through comparing transcriptional profilings of ETO sensitive versus resistant tumor cell lines, the authors found that BCL6 was selectively upregulated in ETO-resistant tumor cells, and their further in vitro and in vivo data suggest that Bcl6 upregulation via the IFN-STAT1-Bcl6 axis conferred tumor resistance to genotoxic stress, and targeting Bcl6 significantly improved therapeutic efficacy of ETO/ADR in mouse tumor models.
Their findings are interesting and may inspire new combinational therapeutic strategy in treating chemotherapy resistant cancers, although a number of issues remain to be further clarified.
Major concerns:
- Through using in vitro assays, the authors defined a panel of …
Author Response:
Reviewer #2 (Public Review):
In the paper entitled "The Oncoprotein BCL6 Enables Cancer Cells to Evade Genotoxic Stress", through comparing transcriptional profilings of ETO sensitive versus resistant tumor cell lines, the authors found that BCL6 was selectively upregulated in ETO-resistant tumor cells, and their further in vitro and in vivo data suggest that Bcl6 upregulation via the IFN-STAT1-Bcl6 axis conferred tumor resistance to genotoxic stress, and targeting Bcl6 significantly improved therapeutic efficacy of ETO/ADR in mouse tumor models.
Their findings are interesting and may inspire new combinational therapeutic strategy in treating chemotherapy resistant cancers, although a number of issues remain to be further clarified.
Major concerns:
- Through using in vitro assays, the authors defined a panel of genotoxic agents (ETO, ADR, etc) resistant or sensitive tumor cell lines, and indicated the resistance was caused by BCL6 upregulation. It was expected in the following on animal studies, the authors would choose tumor cell lines with clearly defined phenotypes characterized in their study. But it was not the cases in their studies. For examples, in Fig S2C and Fig 7B, the authors used an ambiguous tumor cell line HCT116 to test ETO resistance, which had only a borderline level of resistance to ETO (Fig 1A) but yet sensitive to ADR (Fig S1A), whereas in Fig 2H, the authors chose a tumor cell line (MCF7) not examined in their study, instead of the high ETO-resistant tumor cell lines H661/Capan-2 or high ADR-resistant cell lines DLD-1/H836.
We thank the reviewer very much for these insightful comments.
(1) We sincerely agree with the reviewer that our experiments should be carried out using cell lines that possess clear and potent resistance phenotype. However, some resistant cell lines (e.g., H661 and Capan-2) are hard to form tumors in mice according to published literature or our experiences. That’s why we initially chose the resistant cell line HCT116 for animal studies. To follow the reviewer’s suggestion and further validate our findings, in our revised manuscript, we additionally set up a tumor xenograft mouse model using PANC28 cells that are more resistant to etoposide than HCT116 cells. Our new data consistently showed that the BCL6 abundance in PANC28 xenografts was apparently increased by etoposide treatment, and as expected, BCL6 knockdown significantly sensitized etoposide. We have supplemented these new data in Figure 2D, Figure 2-figure supplement 1C and Figure 7C of our revised manuscript.
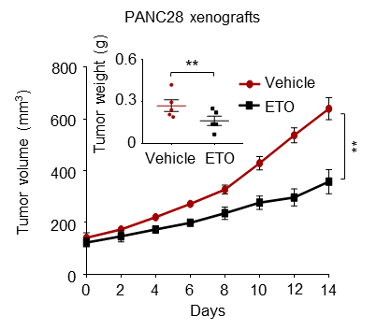
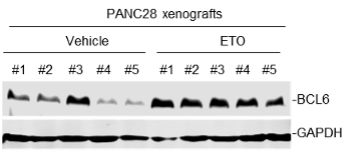
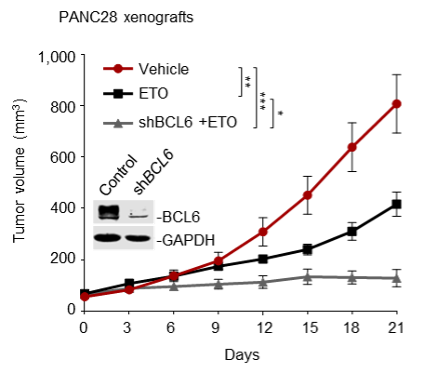
(2) Moreover, we also tested the in vitro sensitizing effects of BCL6 knockdown to etoposide and doxorubicin using Capan-2 and H838 cells that are much more resistant to genotoxic agents. As expected, our results showed that BCL6 genetic knockdown attenuated the clonogenic growth of these cells in the presence of etoposide or doxorubicin. We are sorry that we can't supplement all these figures in our revised manuscript due to limited space. We have added the Capan-2 data in our revised manuscript (Figure 2E).
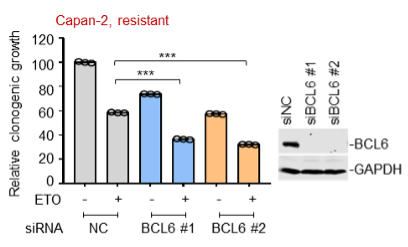
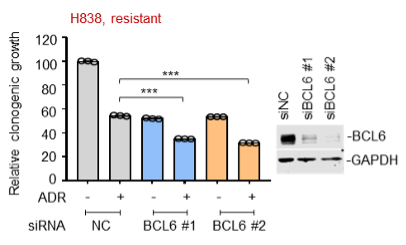
(3) In the previous version of our manuscript, we analyzed published datasets (Biomed Pharmacother. 2014 May;68(4):447-53; PLoS One. 2012;7(9):e45268), and found that BCL6 upregulation was also observed in cells with acquired chemoresistance (MCF7/ETO and A2780/ADR; Figure 1E). We further examined the inhibitory action of BCL6 silencing in the acquired chemo-resistant MCF7/ADR cells that we generated previously in our laboratory. Our results showed that BCL6 interference was sufficient to suppress the growth of MCF7/ADR cells. In attempting to make consistency of used cell lines across the experimental panels in our study, nevertheless, we decided to remove the MCF7/ADR proliferation data in our revised manuscript.
- Fig 3, the concept of tumor cell expressing IFNa/IFNg conferring genotoxic resistance sounds very interesting and novel, but the authors only tested IFNa/g expression at transcriptional level, protein expression data should be also provided.
We appreciate the reviewer’s comments.
In our study, we have examined the protein contents of IFN-α and IFN-γ using an ELISA assay. Our results showed that etoposide treatment led to a significant increase in IFN-α and IFN-γ contents in resistant cells. The results were expressed as fold change over the untreated control (Figure 3, H-I). We have revised the related figure legends to make it clearer to readers.
- Fig 3F-3I, ETO-induced interferon response should be examined comprehensively in different tumor cell lines as listed in Fig 1A/2A. Similarly, effect of exogenous IFNa/IFNg on ETO-resistance should be also examined comprehensively in both sensitive or resistant tumor cell lines. In addition, the effect of blocking IFNg/IFNa on ETO-resistance should be also tested in different tumor cell lines. These data are extremely useful for extending or strengthening the broad impact or influence of their findings.
We appreciate for the reviewer’s suggestion.
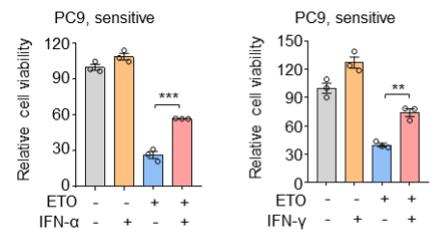
We agree that more cell lines should be examined in the context of exogenous addition of IFNα/IFNγ or IFNα/IFNγ blockade. However, it is hard for us to test all the cell lines as listed in Figure 1A/2A. In our revised manuscript, we expanded cell line panel in this part and supplemented several new data as listed below.
(1) In addition to the sensitive cell line H522 that has been already shown in our previous manuscript, we further tested PC9 cells and consistently found that exogenous addition of IFN-α and IFN-γ also protected PC9 cells from etoposide-induced cell death.
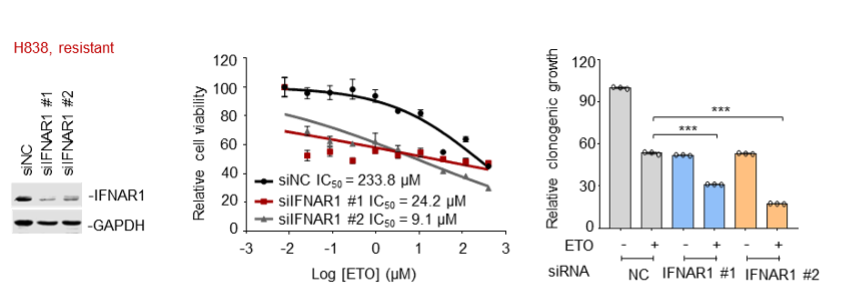
(2) In addition to the resistant cell line Capan-2 that has been already shown in our previous manuscript, we further tested H838 cells and consistently found that knockdown of the IFN-α receptor IFNAR1 led to an enhanced sensitivity of H838 cells to etoposide, as indicated by decreased IC50 values of etoposide and impaired clonogenic growth of H838 cells compared with the control group.
(3) In addition to the resistant cell line PANC28 that has been already shown in our previous manuscript, we further employed Capan-2 and H838 cells and consistently found that antibodies against IFN-γ also increased the killing ability of etoposide towards these resistant cells.
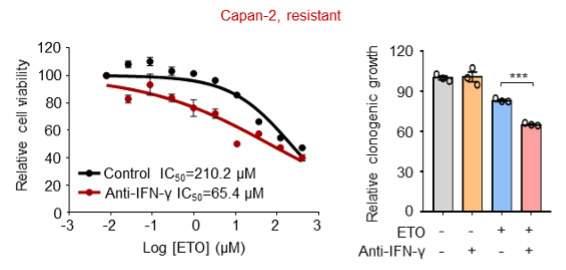
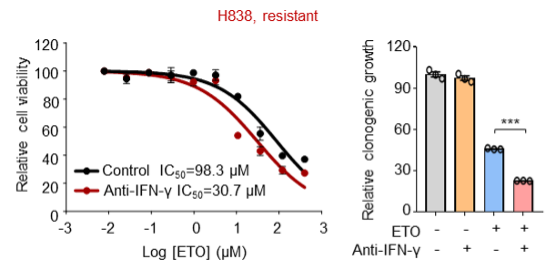
We are sorry that we can't supplement all these figures in our revised manuscript due to limited space. We have added the Capan-2 data in our revised manuscript (Figure 3O and Figure 3-figure supplement 1F).
- Fig 4A-L, the authors examined activation of IFN-STAT1-Bcl6 axis in tumor cells in different angles via different approaches, but using different tumor cell lines in different panels of experiments, making it quite annoying and difficult to judge their findings across different tumor cell lines. At least, ETO or IFNa/IFNg induced STAT1 upregulation and its phosphorylation should be examined comprehensively in both resistant and sensitive tumor cell lines.
We thank so much for this helpful comment.
We are so sorry for the inconsistency of cell lines used in our previous manuscript. We have employed consistent cell lines across the experimental panels and supplemented additional data in our revised manuscript. We chose the chemo-resistant cell line Capan-2, PANC28, H838 and HCT116 for mechanistic studies, and correspondingly, we employed the chemo-sensitive cell line H522, PC9 and PANC-1 for comparison in certain assays.
As suggested by the reviewer, we tested more cell lines to further elucidate the IFN-STAT1-Bcl6 axis. Our results showed that etoposide treatment promoted STAT1 abundance and its phosphorylated levels in etoposide-resistant Capan-2, PANC28 and H838 cells, but not in sensitive H522, PC9 and PANC-1 cells. Additionally, IFN-α and IFN-γ significantly led to a simultaneous increase in STAT1, phosphorylated STAT1 and BCL6 expression in the same resistant cell panel.
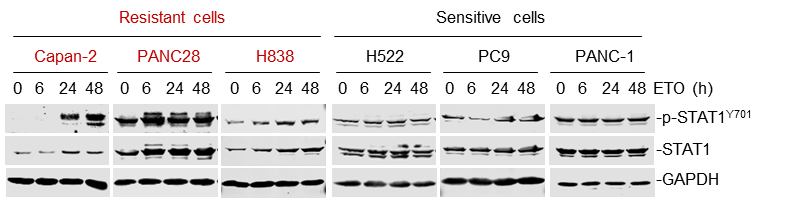
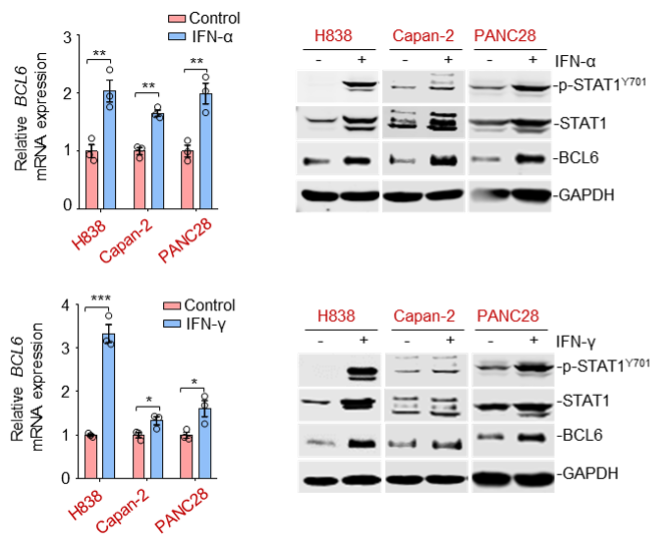
We have supplemented the new data in Figure 4A and Figure 4, C-F of our revised manuscript.
-
Evaluation Summary:
This research explored the role that BCL6 exerts on chemotherapy resistance in solid tumors other than previously reported hematological malignancies. The study also implicates the IFN-BCL6-PTEN axis as an antagonism target for overcoming resistance. This paper will be interesting to scientists who are specialists in cancer chemoresistance and oncological pharmacology.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #3 agreed to share their name with the authors.)
-
Reviewer #1 (Public Review):
This study confirmed the role of BCL6 in tumor cells escaping cytotoxic drugs in vitro. The upstream and downstream molecules of BCL6 were determined by in vitro experiments. The role of BCL6 inhibitor in chemo-sensitization was verified by in vitro experiments and animal experiments,which proved certain clinical application value. These findings establish a rationale for targeting BCL6 to conquer resistance to genotoxic stress in solid tumors which has certain clinical value. However, ample researches have reported during the past years, innovation of this manuscript is required to be addressed. The authors have done a lot of research and obtained abundant data which provides strong support for the research conclusion.
-
Reviewer #2 (Public Review):
In the paper entitled "The Oncoprotein BCL6 Enables Cancer Cells to Evade Genotoxic Stress", through comparing transcriptional profilings of ETO sensitive versus resistant tumor cell lines, the authors found that BCL6 was selectively upregulated in ETO-resistant tumor cells, and their further in vitro and in vivo data suggest that Bcl6 upregulation via the IFN-STAT1-Bcl6 axis conferred tumor resistance to genotoxic stress, and targeting Bcl6 significantly improved therapeutic efficacy of ETO/ADR in mouse tumor models.
Their findings are interesting and may inspire new combinational therapeutic strategy in treating chemotherapy resistant cancers, although a number of issues remain to be further clarified.
Major concerns:
1. Through using in vitro assays, the authors defined a panel of genotoxic agents (ETO, …
Reviewer #2 (Public Review):
In the paper entitled "The Oncoprotein BCL6 Enables Cancer Cells to Evade Genotoxic Stress", through comparing transcriptional profilings of ETO sensitive versus resistant tumor cell lines, the authors found that BCL6 was selectively upregulated in ETO-resistant tumor cells, and their further in vitro and in vivo data suggest that Bcl6 upregulation via the IFN-STAT1-Bcl6 axis conferred tumor resistance to genotoxic stress, and targeting Bcl6 significantly improved therapeutic efficacy of ETO/ADR in mouse tumor models.
Their findings are interesting and may inspire new combinational therapeutic strategy in treating chemotherapy resistant cancers, although a number of issues remain to be further clarified.
Major concerns:
1. Through using in vitro assays, the authors defined a panel of genotoxic agents (ETO, ADR, etc) resistant or sensitive tumor cell lines, and indicated the resistance was caused by BCL6 upregulation. It was expected in the following on animal studies, the authors would choose tumor cell lines with clearly defined phenotypes characterized in their study. But it was not the cases in their studies. For examples, in Fig S2C and Fig 7B, the authors used an ambiguous tumor cell line HCT116 to test ETO resistance, which had only a borderline level of resistance to ETO (Fig 1A) but yet sensitive to ADR (Fig S1A), whereas in Fig 2H, the authors chose a tumor cell line (MCF7) not examined in their study, instead of the high ETO-resistant tumor cell lines H661/Capan-2 or high ADR-resistant cell lines DLD-1/H836.
2. Fig 3, the concept of tumor cell expressing IFNa/IFNg conferring genotoxic resistance sounds very interesting and novel, but the authors only tested IFNa/g expression at transcriptional level, protein expression data should be also provided.
3. Fig 3F-3I, ETO-induced interferon response should be examined comprehensively in different tumor cell lines as listed in Fig 1A/2A. Similarly, effect of exogenous IFNa/IFNg on ETO-resistance should be also examined comprehensively in both sensitive or resistant tumor cell lines. In addition, the effect of blocking IFNg/IFNa on ETO-resistance should be also tested in different tumor cell lines. These data are extremely useful for extending or strengthening the broad impact or influence of their findings.
4. Fig 4A-L, the authors examined activation of IFN-STAT1-Bcl6 axis in tumor cells in different angles via different approaches, but using different tumor cell lines in different panels of experiments, making it quite annoying and difficult to judge their findings across different tumor cell lines. At least, ETO or IFNa/IFNg induced STAT1 upregulation and its phosphorylation should be examined comprehensively in both resistant and sensitive tumor cell lines.
-
Reviewer #3 (Public Review):
Overcoming intrinsic and acquired drug resistance is a major challenge in treating cancer patients because chemoresistance causes recurrence, cancer dissemination and death. In this manuscript, Liu et al. reveals oncoprotein B cell lymphoma 6 (BCL6) as a core component that confers tumor adaptive resistance to genotoxic stress. They show that transactivation of BCL6 by genotoxic agents is associated with a poorer therapeutic efficacy and clinical outcome. Genotoxic agents lead to the transcriptional reprogramming of pro-inflammatory cytokines, which subsequently upregulates BCL6 expression. Then, BCL6 represses PTEN and consequently leads to drug resistant. Accordingly, the BCL6 inhibition enhances etoposide-triggered DNA damage and apoptosis. The conclusions of this paper are mostly well supported by data, …
Reviewer #3 (Public Review):
Overcoming intrinsic and acquired drug resistance is a major challenge in treating cancer patients because chemoresistance causes recurrence, cancer dissemination and death. In this manuscript, Liu et al. reveals oncoprotein B cell lymphoma 6 (BCL6) as a core component that confers tumor adaptive resistance to genotoxic stress. They show that transactivation of BCL6 by genotoxic agents is associated with a poorer therapeutic efficacy and clinical outcome. Genotoxic agents lead to the transcriptional reprogramming of pro-inflammatory cytokines, which subsequently upregulates BCL6 expression. Then, BCL6 represses PTEN and consequently leads to drug resistant. Accordingly, the BCL6 inhibition enhances etoposide-triggered DNA damage and apoptosis. The conclusions of this paper are mostly well supported by data, but some aspects of image acquisition and data analysis need to be clarified and extended.
-
