Mitochondrial genome sequencing of marine leukaemias reveals cancer contagion between clam species in the Seas of Southern Europe
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
This paper is of broad interest to biologists and oncologists who study tumour evolution. The study provides new insights into the propagation of a transmissible cancer in clams. Remarkably, based on the analysis of mitochondrial DNA, the transmissible cancer seems to have jumped species. The findings reported have implications to understand the conditions that allow this cancer to spread across huge regions, threatening certain clam species.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1, Reviewer #2 and Reviewer #3 agreed to share their name with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Clonally transmissible cancers are tumour lineages that are transmitted between individuals via the transfer of living cancer cells. In marine bivalves, leukaemia-like transmissible cancers, called hemic neoplasia (HN), have demonstrated the ability to infect individuals from different species. We performed whole-genome sequencing in eight warty venus clams that were diagnosed with HN, from two sampling points located more than 1000 nautical miles away in the Atlantic Ocean and the Mediterranean Sea Coasts of Spain. Mitochondrial genome sequencing analysis from neoplastic animals revealed the coexistence of haplotypes from two different clam species. Phylogenies estimated from mitochondrial and nuclear markers confirmed this leukaemia originated in striped venus clams and later transmitted to clams of the species warty venus, in which it survives as a contagious cancer. The analysis of mitochondrial and nuclear gene sequences supports all studied tumours belong to a single neoplastic lineage that spreads in the Seas of Southern Europe.
Article activity feed
-

-

Author Response:
Reviewer #1 (Public Review):
Garcia-Souto, Bruzos, and Diaz et al. analyzed hemic neoplasia in warty venus clams at multiple sites throughout Europe. They identified cases of disease in two locations, in Galicia and in the Mediterranean. They then use Illumina sequencing to discover that the samples with cancer DNA had reads which mapped to the mtDNA reference sequences from a different clam species in the same family, suggesting a cross-species transmissible cancer. By mapping reads to both the V. verrucosa and C. gallina mitogenomes they showed that more reads mapped to C. gallina in cancer samples compared to matched host tissue samples, and this was consistent across the whole mitogenome. Phylogenetic analysis of mtDNA genes of the host and cancer samples as well as identification of SNVs at a short region of one …
Author Response:
Reviewer #1 (Public Review):
Garcia-Souto, Bruzos, and Diaz et al. analyzed hemic neoplasia in warty venus clams at multiple sites throughout Europe. They identified cases of disease in two locations, in Galicia and in the Mediterranean. They then use Illumina sequencing to discover that the samples with cancer DNA had reads which mapped to the mtDNA reference sequences from a different clam species in the same family, suggesting a cross-species transmissible cancer. By mapping reads to both the V. verrucosa and C. gallina mitogenomes they showed that more reads mapped to C. gallina in cancer samples compared to matched host tissue samples, and this was consistent across the whole mitogenome. Phylogenetic analysis of mtDNA genes of the host and cancer samples as well as identification of SNVs at a short region of one single-copy nuclear locus suggest that all cancer samples come from a single C. gallina transmissible cancer clone. All data agree that a single lineage of cancer from C. gallina is responsible for all identified cancers in V. verrucosa.
There are a few sections where there are either unclear methods or the methods do not quite match the descriptions of the results.
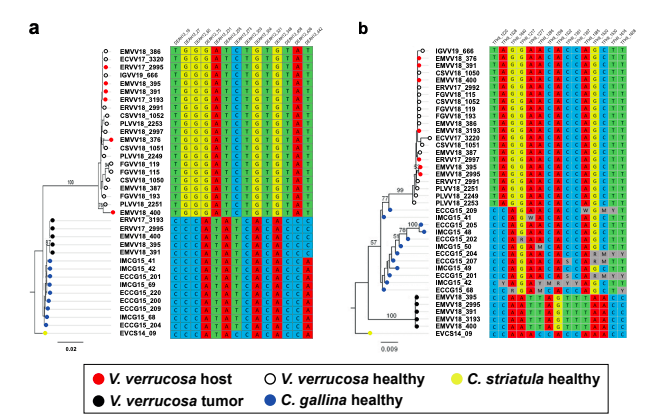
- Regarding mapping of reads to different reference Cox1 sequences (for Figure 2a): "Then, we mapped the paired-end reads onto a dataset containing non-redundant mitochondrial Cytochrome C Oxidase subunit 1 (Cox1) gene references from 137 Vererid clam species." I do not see where this is explained anywhere in the methods, where this list of references comes from, or what is in it.
Answer: We retrieved a dataset of 3,745 sequences comprising all the barcode-identified venerid clam Cox1 fragments available from the Barcode of Life Data System (BOLD, http://www.boldsystemns.org/). Redundancy was removed using CD-HIT (Fu, et al. 2012), applying a cut-off of 0.9 sequence identity, and sequences were trimmed to cover the same region. Whole-genome sequencing data from both healthy and tumoral warty venus clams was mapped onto this dataset, containing 118 venerid species-unique sequences, using BWA-mem, filtering out reads with mapping quality below 60 (-q60) and quantifying the overall coverage for each sequence with samtools idxstats. PCR primers were designed with Primer3 v2.3.7 (Koressaar, et al. 2018) to amplify a fragment of 354 bp from the Cox1 mitochondrial gene of V. verrucosa and C. gallina (F: CCT ATA ATA ATT GGK GGA TTT GG, R: CCT ATA ATA ATT GGK GGA TTT GG). PCR products were purified with ExoSAP-IT and sequenced by Sanger sequencing.
Action: We have included this new information in the methods section.
- Regarding de novo assembly of mitogenomes: "Hence, we employed bioinformatic tools to reconstruct the full mitochondrial DNA (mtDNA) genomes in representative animals from the two species involved....Then, we mapped the paired-end sequencing data from the six neoplastic specimens with evidence of interspecies cancer transmission onto the two reconstructed species-specific mtDNA genomes." In contrast to this, the methods say, "Then, we run MITObim v1.9.1 (Hahn, Bachmann, & Chevreux, 2013) to assemble the full mitochondrial genome of all sequenced samples, using gene baits from the following Cox1 and 16S reference genes to prime the assembly of clam mitochondrial genomes." It is unclear which method was used.
Answer: In total, we performed whole-genome sequencing on 23 samples from 16 clam specimens, which includes eight neoplastic and eight non-neoplastic animals by Illumina pairedend libraries of 350 bp insert size and reads 150 bp long. First we assembled the mitochondrial genomes of one V. verrucosa (FGVV18_193), one C. gallina (ECCG15_201) and one C. striatula (EVCS14_02) specimens with MITObim v1.9.1 (Hahn, et al. 2013), using gene baits from the 7 following Cox1 and 16S reference genes to prime the assembly of clam mitochondrial genomes: V. verrucosa (Cox1, with GenBank accession number KC429139; and 16S: C429301), C. gallina (Cox1: KY547757, 16S: KY547777) and C. striatula (Cox1: KY547747, 16S: KY547767). These draft sequences were polished twice with Pilon v1.23 (Walker, et al. 2014), and conflictive repetitive fragments from the mitochondrial control region were resolved using long read sequencing with Oxford Nanopore technologies (ONT) on a set of representative samples from each species and tumours. ONT reads were assembled with Miniasm v0.3 (Li 2016) and corrected using Racon v1.3.1 (Vaser, et al. 2017). Protein-coding genes, rDNAs and tRNAs were annotated on the curated mitochondrial genomes using MITOS2 web server (Bernt, et al. 2013), and manually curated to fit ORFs as predicted by ORF-FINDER (Rombel, et al. 2002). Then, we employed the entire mitochondrial DNAs of V. verrucosa (FGVV18_193) and C. gallina (ECCG15_201) as “references” to map reads from individuals with neoplasia, filter reads matching either mitogenome and assemble and polish their two (healthy and tumoral) mitogenomes individually as above. Further healthy individuals were later sequenced and their mitogenomes assembled, to further investigate the geographic and taxonomic spread of this neoplasia.
Action: We have included this information in the methods section (page 21-22), and in the results (pages 7 and 8). mtDNA annotations are now shown in Supplementary Figure 3. Nucleotide data for the mitochondrial DNA assemblies has been uploaded to GenBank under accession numbers MW662590-MW662611 and will be released upon publication or request.
There is one minor claim which may not be fully supported by the data: the statement that, "The analysis of mitochondrial and nuclear gene sequences revealed no nucleotide divergence between the seven tumours sequenced." If I am understanding the filtering of the SNVs from the nuclear gene correctly, only the presence or absence of the 14 SNVs that were fixed within each of the two species were analyzed. Therefore, it is unclear whether the authors looked for any additional somatic mutations within the cancer lineage that would have occurred at other positions. For mitochondria, the authors state that sequences were "extracted from paired-end sequencing data," but it is not explained how this was done. The data suggest that there are no differences between cancer samples in the 13 coding genes and 2 rDNA genes, but data on possible SNVs in the intergenic regions is not shown.
Answer: We obtained a preliminary nuclear assembly using short-reads only. Obviously, the resulting assemblies are fragmented and incomplete. This has limited the identification of candidate regions shared by the three genomes (V. verrucosa and both Chamelea clams). Out of the 44 candidate nuclear fragments we tested, only two (DEAH12 and TFHII) turned out to give good PCR products, adequate for Sanger sequencing. As mentioned above, we now provide additional data on a second gene (TFIIH), identified and selected on the same basis as DEAH12. We find 14 and 15 sites, respectively, for the DEAH12 and the TFIIH loci, with fixed SNVs (allele frequency >95%) that allowed to discriminate between the three relevant species (V. verrucosa, C. gallina and C. striatula) and the tumour. These diagnostic nucleotides were then used to filter the reads from individuals with neoplasia harbouring both DNA’s. Variation within the host lineage but not within the tumour was found along the nuclear DNA fragments employen in the ML phylogenies (see figure below).
<img src= https://cdn.elifesciences.org/public-review-media/public-reviews/PAhXWiM.png.png" />
Figure. Molecular phylogenies based on the two selected nuclear markers. (a) DEAH12 gene and (b) TFIIH gene, and diagnostic loci discriminating among species and tumour. Bootstrap support values (500 replicates) from ML analyses above 50 are shown above the corresponding branches. Note all diagnostic nucleotides are identical between tumours (black dots).
Regarding the mtDNA, firstly, we assembled the mitochondrial genomes of one V. verrucosa (FGVV18_193), one C. gallina (ECCG15_201) and one C. striatula (EVCS14_02) specimens with MITObim v1.9.1 (Hahn, et al. 2013). Then, we employed the entire mitochondrial DNAs from V. verrucosa (FGVV18_193) and C. gallina (ECCG15_201) as “references” to map reads from individuals with neoplasia, filter reads matching either mitogenome and assemble and polish their two (healthy and tumoral) mitogenomes individually as above. Further healthy individuals were later sequenced and their mitogenomes assembled, to further investigate the geographic and taxonomic spread of this neoplasia. Despite the usefulness of the mitochondrial control region (CR) to detect differences among lineages, we refrained from using it for two reasons. (1) The CR shows considerable variation in both length and sequence among the three species, making their alignment difficult (in fact, previous phylogenetic studies based on whole mitochondrial DNA sequences in Veneridae excluded the CR: https://doi.org/10.1111/zsc.12454), and (2) the CR contains quasi-but-not-identical tandem repeats, as a other mollusks (i.e., the Venerid Dosinia clams https://doi.org/10.1371/journal.pone.0196466 or the Littorina marine snails https://doi.org/10.1016/j.margen.2016.10.006). In our case, repeats are larger than the short-reads insert size, and even though we could infer them by means of long read sequencing, polishing the resulting consensus sequences to overcome the intrinsic error rate of those lectures would yield inconclusive results, hindering the comparison between normal and tumoral haplotypes.
Action: We updated the methods for the mitochondrial DNA analyses (pages 21-22, 24) and the nuclear DNA analyses (page 23). We now include new data in the results and discussion (pages 9-10).
Reviewer #2 (Public Review):
In rare but well-documented instances, certain types of cancers can transmit horizontally. These transmissible cancers have a clonal origin and have adapted to bypass allorecognition. A form of marine leukemia (hemic neoplasia or HM) belongs to this class of transmissible cancers and has been detected in several bivalve species (oysters, mussels, cockles and clams). Although HM mostly propagates within the same bivalve species, instances of cross-species transmission have been reported. To better understand the mode of transmission of HM, Garcia-Souto et al. analysed mitochondrial DNA (mtDNA) by next generation sequencing in different bivalve species collected in the Mediterranean Sea and the Atlantic Ocean. The authors found that HM isolated in Venus verrucosa contained mtDNA that actually matched Chamelea gallina. Analysis of the nuclear gene DEAH12 also showed single nucleotide polymorphisms (SNPs) matching C. gallina DNA. Based on mtDNA and DEAH12 sequences, the authors use Bayesian inference to generate phylogenetic trees showing that HM found in V. verrucosa is much closer to C. gallina than the host species. They conclude that HM propagated from C. gallina to V. verrucosa.
Overall, the study is well performed with enough samples analysed. The results are quite convincing but there are also some concerns.
- Transmissible cancers are known to split into clades based on mtDNA differential rate of evolution and also to incorporate mtDNA from exogenous sources, so one has to be extra careful that the results prove cross-species transmission and not HM divergence into two clades and/or exogenous acquisition. Samples HM ERVV17-2997 and EMVV18-376, both at the N1 stage, appear devoid of C. gallinae mtDNA and do not appear to have been screened for DEAH12. One explanation for this result is that there are too few HM cells in the samples (but supplementary Figure 1 shows some HM cells in ERVV17-2997. However, a different explanation is that these samples contain V. verrucosae mtDNA. ERVV17-2997 and EMVV18-376 could have been analysed in greater depth to verify that they also contained C. gallinae mtDNA and typical DEAH12 SNPs.
Answer: Despite the high sequencing coverage obtained for the sequenced individuals, we did not find foreign reads in the N1 tumours (ERVV17-2997 and EMVV18-73) to mitochondrial nor nuclear (i.e., DEAH12, TFHII) level. This is most likely due to a very low proportion of neoplastic cells in their tissues.
Action: We have added a sentence on page 8 that discuss this issue.
- To strengthen their argument, the authors could have analysed a few more nuclear genes for specific SNPs, although the sensitivity of this approach will depend on the depth of sequencing.
Answer: We obtained a preliminary nuclear assembly using short-reads only. Obviously, the resulting assemblies are fragmented and incomplete. This has limited the identification of candidate regions shared by the three genomes (V. verrucosa and both Chamelea clams). Out of the 44 candidate nuclear fragments we tested, only two (DEAH12 and TFHII) turned out to give good PCR products, adequate for Sanger sequencing. As mentioned above, we now provide additional data on a second gene (TFIIH), identified and selected on the same basis as DEAH12. Individual ML phylogenies for these two fragments evidenced that tumours cluster together and separately from the host species and, in the case of DEAH12, closer to C. gallina. The MSC phylogeny was rebuilt including this new nuclear fragment. 12 In addition, we conducted a comparative screening of tandem repeats on the genomes of C. gallina and V. verrucosa. Two DNA satellites, namely CL4 and CL17, of, respectively, 332 and 429 bp monomer size, were very abundant in C. gallina and in the tumoral animals, but absent from all healthy V. verrucosa specimens. FISH probes designed for these satellites mapped on the heterochromatic regions, mainly in subcentromeric and subtelomeric positions, of both C. gallina and the neoplastic metaphases found in V. verrucosa, but were absent from the normal metaphases of the host species V. verrucosa. These results were consistent with the genomic abundance of these satellites in the NGS data and strongly suggest that these chromosomes derive from C. gallina.
Action: We include the analysis of one additional nuclear locus, TFIIH (pages 9-10). We have obtained new ML and MSC phylogenies including this new locus (pages 9-10, figures 3b-c). Additional FISH approach looking for satellite DNA CL4 and CL11 was performed (page 10, figure 3d, supplementary figure 5). The methods section has been updated accordingly (pages 20- 21, 23-24).
- It would have been interesting to have more information in the Discussion on the potential immunological barriers that this tumour needs to overcome for cross-species transmission.
Answer: At a glance, we could argue/discuss that this transmissibility, inside or cross-species, is prone to occur in bivalves due to their filtering feeding system and the fact that their immune system is not entirely developed and yet to be completely understood, as the reviewer may know. Also, it would be tempting to suggest that some genetic restrictions allowing for cancer contagion happening only between close taxa might be in place, but, unfortunately we do not have the means to state that with our current data.
Action: At this point, no specific action has been taken for this query. However, we are happy to include something in the discussion if the reviewer still thinks this is relevant for improving the manuscript.
-
Evaluation Summary:
This paper is of broad interest to biologists and oncologists who study tumour evolution. The study provides new insights into the propagation of a transmissible cancer in clams. Remarkably, based on the analysis of mitochondrial DNA, the transmissible cancer seems to have jumped species. The findings reported have implications to understand the conditions that allow this cancer to spread across huge regions, threatening certain clam species.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1, Reviewer #2 and Reviewer #3 agreed to share their name with the authors.)
-
Reviewer #1 (Public Review):
Garcia-Souto, Bruzos, and Diaz et al. analyzed hemic neoplasia in warty venus clams at multiple sites throughout Europe. They identified cases of disease in two locations, in Galicia and in the Mediterranean. They then use Illumina sequencing to discover that the samples with cancer DNA had reads which mapped to the mtDNA reference sequences from a different clam species in the same family, suggesting a cross-species transmissible cancer. By mapping reads to both the V. verrucosa and C. gallina mitogenomes they showed that more reads mapped to C. gallina in cancer samples compared to matched host tissue samples, and this was consistent across the whole mitogenome. Phylogenetic analysis of mtDNA genes of the host and cancer samples as well as identification of SNVs at a short region of one single-copy nuclear …
Reviewer #1 (Public Review):
Garcia-Souto, Bruzos, and Diaz et al. analyzed hemic neoplasia in warty venus clams at multiple sites throughout Europe. They identified cases of disease in two locations, in Galicia and in the Mediterranean. They then use Illumina sequencing to discover that the samples with cancer DNA had reads which mapped to the mtDNA reference sequences from a different clam species in the same family, suggesting a cross-species transmissible cancer. By mapping reads to both the V. verrucosa and C. gallina mitogenomes they showed that more reads mapped to C. gallina in cancer samples compared to matched host tissue samples, and this was consistent across the whole mitogenome. Phylogenetic analysis of mtDNA genes of the host and cancer samples as well as identification of SNVs at a short region of one single-copy nuclear locus suggest that all cancer samples come from a single C. gallina transmissible cancer clone. All data agree that a single lineage of cancer from C. gallina is responsible for all identified cancers in V. verrucosa.
There are a few sections where there are either unclear methods or the methods do not quite match the descriptions of the results.
1. Regarding mapping of reads to different reference Cox1 sequences (for Figure 2a): "Then, we mapped the paired-end reads onto a dataset containing non-redundant mitochondrial Cytochrome C Oxidase subunit 1 (Cox1) gene references from 137 Vererid clam species." I do not see where this is explained anywhere in the methods, where this list of references comes from, or what is in it.
2. Regarding de novo assembly of mitogenomes: "Hence, we employed bioinformatic tools to reconstruct the full mitochondrial DNA (mtDNA) genomes in representative animals from the two species involved....Then, we mapped the paired-end sequencing data from the six neoplastic specimens with evidence of interspecies cancer transmission onto the two reconstructed species-specific mtDNA genomes." In contrast to this, the methods say, "Then, we run MITObim v1.9.1 (Hahn, Bachmann, & Chevreux, 2013) to assemble the full mitochondrial genome of all sequenced samples, using gene baits from the following Cox1 and 16S reference genes to prime the assembly of clam mitochondrial genomes." It is unclear which method was used.There is one minor claim which may not be fully supported by the data: the statement that, "The analysis of mitochondrial and nuclear gene sequences revealed no nucleotide divergence between the seven tumours sequenced." If I am understanding the filtering of the SNVs from the nuclear gene correctly, only the presence or absence of the 14 SNVs that were fixed within each of the two species were analyzed. Therefore, it is unclear whether the authors looked for any additional somatic mutations within the cancer lineage that would have occurred at other positions. For mitochondria, the authors state that sequences were "extracted from paired-end sequencing data," but it is not explained how this was done. The data suggest that there are no differences between cancer samples in the 13 coding genes and 2 rDNA genes, but data on possible SNVs in the intergenic regions is not shown.
-
Reviewer #2 (Public Review):
In rare but well-documented instances, certain types of cancers can transmit horizontally. These transmissible cancers have a clonal origin and have adapted to bypass allorecognition. A form of marine leukemia (hemic neoplasia or HM) belongs to this class of transmissible cancers and has been detected in several bivalve species (oysters, mussels, cockles and clams). Although HM mostly propagates within the same bivalve species, instances of cross-species transmission have been reported. To better understand the mode of transmission of HM, Garcia-Souto et al. analysed mitochondrial DNA (mtDNA) by next generation sequencing in different bivalve species collected in the Mediterranean Sea and the Atlantic Ocean. The authors found that HM isolated in Venus verrucosa contained mtDNA that actually matched Chamelea …
Reviewer #2 (Public Review):
In rare but well-documented instances, certain types of cancers can transmit horizontally. These transmissible cancers have a clonal origin and have adapted to bypass allorecognition. A form of marine leukemia (hemic neoplasia or HM) belongs to this class of transmissible cancers and has been detected in several bivalve species (oysters, mussels, cockles and clams). Although HM mostly propagates within the same bivalve species, instances of cross-species transmission have been reported. To better understand the mode of transmission of HM, Garcia-Souto et al. analysed mitochondrial DNA (mtDNA) by next generation sequencing in different bivalve species collected in the Mediterranean Sea and the Atlantic Ocean. The authors found that HM isolated in Venus verrucosa contained mtDNA that actually matched Chamelea gallina. Analysis of the nuclear gene DEAH12 also showed single nucleotide polymorphisms (SNPs) matching C. gallina DNA. Based on mtDNA and DEAH12 sequences, the authors use Bayesian inference to generate phylogenetic trees showing that HM found in V. verrucosa is much closer to C. gallina than the host species. They conclude that HM propagated from C. gallina to V. verrucosa.
Overall, the study is well performed with enough samples analysed. The results are quite convincing but there are also some concerns.
1. Transmissible cancers are known to split into clades based on mtDNA differential rate of evolution and also to incorporate mtDNA from exogenous sources, so one has to be extra careful that the results prove cross-species transmission and not HM divergence into two clades and/or exogenous acquisition. Samples HM ERVV17-2997 and EMVV18-376, both at the N1 stage, appear devoid of C. gallinae mtDNA and do not appear to have been screened for DEAH12. One explanation for this result is that there are too few HM cells in the samples (but supplementary Figure 1 shows some HM cells in ERVV17-2997. However, a different explanation is that these samples contain V. verrucosae mtDNA. ERVV17-2997 and EMVV18-376 could have been analysed in greater depth to verify that they also contained C. gallinae mtDNA and typical DEAH12 SNPs.
2. To strengthen their argument, the authors could have analysed a few more nuclear genes for specific SNPs, although the sensitivity of this approach will depend on the depth of sequencing.
3. It would have been interesting to have more information in the Discussion on the potential immunological barriers that this tumour needs to overcome for cross-species transmission.
-
Reviewer #3 (Public Review):
The authors first investigated by cyto-histological examination the prevalence of hemic neoplasia, a leukemia-like disease of bivalves, in 345 warty venus clam V. verrucosa from six sampling regions in the Atlantic and the Mediterranean coasts of Europe. Eight specimens from two sampling points in Spain were diagnosed infected by hemic neoplasia. Electron microscopy and karyotyping confirmed abnormal cells suggestive of neoplasia. They secondly carried out whole-genome sequencing of eight tumoral haemolymphs and host feet. They also sequenced seven genomes of healthy V. verrucosa. Preliminary investigation of mtCOI sequences suggested cancer mitochondria could be related to a sister species, the striped venus C. gallina, and the authors sequenced two genomes of this species together with one genome of its …
Reviewer #3 (Public Review):
The authors first investigated by cyto-histological examination the prevalence of hemic neoplasia, a leukemia-like disease of bivalves, in 345 warty venus clam V. verrucosa from six sampling regions in the Atlantic and the Mediterranean coasts of Europe. Eight specimens from two sampling points in Spain were diagnosed infected by hemic neoplasia. Electron microscopy and karyotyping confirmed abnormal cells suggestive of neoplasia. They secondly carried out whole-genome sequencing of eight tumoral haemolymphs and host feet. They also sequenced seven genomes of healthy V. verrucosa. Preliminary investigation of mtCOI sequences suggested cancer mitochondria could be related to a sister species, the striped venus C. gallina, and the authors sequenced two genomes of this species together with one genome of its sibling species Chamelea striatula. Mitogenome analysis confirmed cancerous clams were infected by the same clonal lineage of transmissible neaoplasia that have emerged in a C. gallina host. The analysis of a nuclear gene, DEAH12, confirmed the result. To find out whether this transmissible cancer lineage was still present in the donor species, the authors conducted histological inspection of 166 striped venus clams but found none with evidence of hemic neoplasia. The donor species could possibly have become resistant to this transmissible neoplasia, or more sampling would be needed to find infected individuals (8/345 versus 0/166, P=0.06).
Overall the study provides convincing evidences of a new transmissible cancer that crossed species boundaries in venus clams. This is an important finding that should make a nice contribution to Elife. My concerns are minor and mostly about methodological issues. I'd mostly like to understand how the authors came with a single nuclear gene while they have high coverage genome data. However, they do not need more data to support their conclusions and it's mostly about clarifying the methodology.
Transmissible cancers are important study systems in many points, somatic evolution, metastasis, self recognition, host-parasite co-evolution, clonal interference, comparative oncology etc.. although they were thought rare exceptions. The field has long remained limited to the two mammals known cases, but bivalve transmissible neoplasia have recently emerged as a new fascinating playground to study the ecology and evolution of transmissible cancers. One new observation is host species shift, as reported here in venus clams. This study comes at an early stage of an emerging research program on unique living entities we know very little about, and will undoubtedly have a strong impact.
-

{kind=link}