An early Sox2-dependent gene expression program required for hippocampal dentate gyrus development
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
The hippocampus is a brain area central for cognition. Mutations in the human SOX2 transcription factor cause neurodevelopmental defects, leading to intellectual disability and seizures, together with hippocampal dysplasia. We generated an allelic series of Sox2 conditional mutations in mouse, deleting Sox2 at different developmental stages. Late Sox2 deletion (from E11.5, via Nestin-Cre) affects only postnatal hippocampal development; earlier deletion (from E10.5, Emx1-Cre) significantly reduces the dentate gyrus, and the earliest deletion (from E9.5, FoxG1-Cre) causes drastic abnormalities, with almost complete absence of the dentate gyrus. We identify a set of functionally interconnected genes (Gli3, Wnt3a, Cxcr4, p73 and Tbr2), known to play essential roles in hippocampal embryogenesis, which are downregulated in early Sox2 mutants, and (Gli3 and Cxcr4) directly controlled by SOX2; their downregulation provides plausible molecular mechanisms contributing to the defect. Electrophysiological studies of the Emx1Cre mouse model reveal altered excitatory transmission in CA1 and CA3 regions.
Article activity feed
-

##Author Response
We thank the editors and the reviewers for a number of useful criticisms and suggestions, and for the opportunity given to us, as authors, to publicly reply to the comments. This is a useful exercise, which brings to the attention of the reader lights, but also shadows of the reviewing process, and that we hope will lead in future to develop a better approach to it. Here, we will reply to a number of selected issues which appear to us to be of particular relevance.
Reviewer 1
Reviewer 1 disqualifies our work altogether, based on her/his statement that: “In the paper by Mercurio et al, the authors examine the role of SOX2 in the development of mouse hippocampal dentate gyrus. Using conditionally mutant SOX2 mice the authors show that early, but not late, deletion of SOX2 leads to developmental impairments of the dentate …
##Author Response
We thank the editors and the reviewers for a number of useful criticisms and suggestions, and for the opportunity given to us, as authors, to publicly reply to the comments. This is a useful exercise, which brings to the attention of the reader lights, but also shadows of the reviewing process, and that we hope will lead in future to develop a better approach to it. Here, we will reply to a number of selected issues which appear to us to be of particular relevance.
Reviewer 1
Reviewer 1 disqualifies our work altogether, based on her/his statement that: “In the paper by Mercurio et al, the authors examine the role of SOX2 in the development of mouse hippocampal dentate gyrus. Using conditionally mutant SOX2 mice the authors show that early, but not late, deletion of SOX2 leads to developmental impairments of the dentate gyrus. A drawback of their study is that these findings have been reported previously by the group (Favaro et al. 2009; Ferri et al. 2013).”
The statement reported in bold is simply not true. In Favaro et al. 2009 (Nat Neurosci 12:1248), we demonstrated that nes-Cre-mediated Sox2 deletion leads to defects in postnatal, but not embryonic, hippocampal neurogenesis. In Ferri et al. 2013 (Development 140:1250), we demonstrated that FoxG1Cre-mediated Sox2 deletion leads to defective development of the VENTRAL forebrain. The presence, at the end of gestation, of hippocampal defects was just mentioned in one sentence: - “the hippocampus, at E18.5, was severely underdeveloped (not shown)” (line 1, page 1253)-, and not analyzed any further. In the present work, we describe in detail, starting from E12.5, up to E18.5, how the hippocampal defect develops, and undertake a detailed study of downstream gene expression and cellular defects arising in mutants.
It is unfortunate that the reviewer further insists on the same misleading, and unfounded statement – see her/his comment 3, highlighted in bold character: “the authors state "...remarkably, in the FoxG1-Cre cKO, the DG appears to be almost absent (Figure 2A).". The question is why this finding is remarkable as it already was published in (Ferri et al. 2013)”. As mentioned above, we only remark, in Ferri et al., that the hippocampus was severely underdeveloped (not shown).
Reviewer 2
Reviewer 2 states, already at the beginning: “I am concerned about a major confounding issue (see below).” ... “The authors rely on Foxg1-Cre for their main evidence that very early deletion of Sox2 leads to near loss of the dentate. However, it doesn't appear that the authors are aware that Foxg1 het mice have a fairly significant dentate phenotype (see this paper).”
The reviewer refers to the fact that, to delete Sox2, we need to express a Cre gene “knocked-in” into the Foxg1 gene; hence, heterozygous and homozygous Sox2 deletions will be accompanied by heterozygous loss of Foxg1. If Foxg1 is important for hippocampus development, the absence of a Foxg1 allele will affect the phenotype.
Unfortunately, the statement of the reviewer is subtly misleading, and leads the reader who has not checked the data reported in the cited paper (Shen et al., 2006) to erroneously believe that heterozygous loss of Foxg1 may be responsible for the effects that we report upon homozygous Sox2 deletion. In contrast to the statement made by the reviewer, the paper cited by the reviewer documents that, while heterozygous loss of Foxg1 leads to important POSTNATAL dentate gyrus abnormalities, the PRENATAL development of the dentate gyrus is essentially normal (Figure 6) (“a subtle and inconsistent defect” of the ventral blade observed in about 50% of the mice at E18.5, according to the authors of that paper). Compare “subtle and inconsistent defect” by Shen et al. with “fairly significant dentate phenotype”, as stated by the reviewer. As our paper is entirely focused on defects seen in PRENATAL development in Foxg1Cre; Sox2 mutants, the subtle and inconsistent defects seen by Shen et al. are in sharp contrast with the deep defects seen in embryonic development in our Foxg1Cre;Sox2-/- mutants, and in agreement with the similarity we observe between wild type and heterozygous Foxg1Cre;Sox2+/- embryos (page 5, lines 140-145, of the version of the Full Submission for publication on August 30). An example showing the comparison between a Wild type, a FoxG1 +/- heterozygote;Sox2+/- heterozygote and a FoxG1 heterozygote;Sox2-/- homozygote is now shown in the accompanying figure.
Obviously the incorrect statement kills our paper by itself. If the reviewer had doubts, we could have provided plenty of additional data demonstrating the lack of significant differences between Foxg1CRE Sox2+/- and wild type (Sox2+/+) embryos, as we stated in our paper.
There is an additional interesting comment by Reviewer 2 (see points 2 and 6). The reviewer argues that “The only two direct targets they find don't seem likely to be important players in the phenotypes they describe”. The Reviewer excludes the Gli3 gene (a direct Sox2 target, see Fig. 6), as a possible important player, in spite of the observation that Gli3 is decreased, at early developmental stages, in the cortical hem (Figure 5). The reviewer says “The Gli3 [mutation] phenotypes that have been published are quite distinct from this”. We object that the Gli3 phenotypes are indeed more severe than the phenotype of our mutant, and include failure to develop a dentate gyrus. However, this observation does not preclude the hypothesis that the decreased expression of Gli3 in our mutant is directly responsible for the phenotype we observe. The more severe phenotype of the Gli3 mutants is in fact due to a germ-line null mutation, whereas, in our Foxg1-Cre Sox2 mutants, we observe only a reduction of Gli3 expression, around E12.5 (Fig. 5), that is compatible with a less severe dentate gyrus phenotype. The Reviewer adds that Wnt3A, based on the phenotype of the knock-out mice, similar to that of our Sox2 deleted mice, is a more relevant gene, but it is not a direct target of Sox2. However, the fact that Wnt3A is apparently not directly regulated by Sox2 is not necessarily to be considered a “minus”; Sox2, being a transcription factor, is expected to directly regulate a multiplicity of genes, whose expression will affect the expression of other genes. Indeed, we presented in Fig 6D the hypothesis that decreased expression of Gli3 may contribute to decreased expression of Wnt3A, as already proposed by Grove et al. (1998) based on the observation that Gli3 null mutants lose the expression of Wnt3A (and other Wnt factors) from the cortical hem. The additional suggestion made by the Reviewer, in the context of the Wnt3A hypothesis, to investigate LEF1, as a potential direct Sox2 target, and its expression, is certainly interesting, but, as stated by the reviewer, LEF1 is downstream to Wnt3A, and, by itself, its hypothetical regulation by Sox2 would not explain the downregulation of Wnt3A. Moreover, we already have evidence that Sox2 does not directly regulate Wnt3A (unpublished).
Reviewer 1 and 2
Both Reviewer 1 and 2 have questions about the timing of Sox2 ablation in the Sox2 mutants obtained with the three different Cre deleters. As we state in the text (pages 4, 6), Foxg1-Cre deletes at E.9.5 (Ferri et al., 2013; Hébert and McConnell, 2000); Emx1-Cre deletes from E10.5 onwards, but not at E9.5 (Gorski et al., 2002; see also Shetty AS et al., PNAS 2013, E4913); Nestin-Cre deletes at later stages, around E12.5 (Favaro et al. 2009).
Reviewer 3
We thank Reviewer 3 for the useful considerations and suggestions, which constructively help to improve the paper.
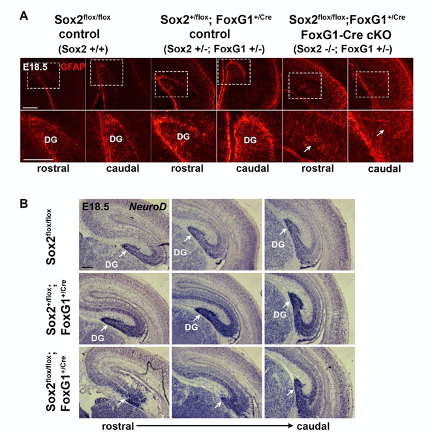
Evidence that Sox2+/-;FoxG1+/- hippocampi at E18.5 do not significantly differ from wild type (Sox2+/+, FoxG1+/+) controls. In contrast, Sox2-/-;FoxG1+/- hippocampi are severely defective. (A) GFAP immunofluorescence at E18.5 on coronal sections of control and FoxG1-Cre cKO hippocampi (controls n=6, mutants n=4). (B) In situ hybridization at E18.5 for NeuroD (controls n=4, mutants n=3) on coronal sections of control and FoxG1-Cre cKO hippocampi. Arrows indicate dentate gyrus (DG); note the strong decrease of the dentate gyrus, and the radial glia (GFAP) disorganization in cKO.
The Sox2flox/flox genotype corresponds to wild type mice (Sox2+/+). The Sox2+/flox ; FoxG1Cre genotype corresponds to Sox2+/-; FoxG1+/- controls. The Sox2flox/flox ; FoxG1Cre genotype corresponds to Sox2-/-; FoxG1+/- mutants. -
###Reviewer #3:
This paper investigates the role of Sox2 in early hippocampal development. Previously the authors investigated conditional knockout mice using a Nestin-Cre line and found few phenotypes. The authors hypothesised that Sox2 may have greater impact on earlier developmental stages. The authors used a similar approach in a previous paper (Ferri et al., 2003) studying the ventral forebrain. To test this in the dorsal telencephalon they generated conditional knockout mice using both Emx1-Cre and FoxG1Cre driver lines. These lines displayed more significant phenotypes in the hippocampus, particularly in the cortical hem and dentate gyrus, and were most severe in the FoxG1Cre cross.
The study is well executed and carefully thought through. Appropriate controls have been included for all experiments.
In Figure 6, the data on Gli3 …
###Reviewer #3:
This paper investigates the role of Sox2 in early hippocampal development. Previously the authors investigated conditional knockout mice using a Nestin-Cre line and found few phenotypes. The authors hypothesised that Sox2 may have greater impact on earlier developmental stages. The authors used a similar approach in a previous paper (Ferri et al., 2003) studying the ventral forebrain. To test this in the dorsal telencephalon they generated conditional knockout mice using both Emx1-Cre and FoxG1Cre driver lines. These lines displayed more significant phenotypes in the hippocampus, particularly in the cortical hem and dentate gyrus, and were most severe in the FoxG1Cre cross.
The study is well executed and carefully thought through. Appropriate controls have been included for all experiments.
In Figure 6, the data on Gli3 has been verified with additional luciferase data. The data on Cxcr4 has been previously published and has not been further verified with luciferase analysis. Including panel C in the figure may not be justified unless additional data is included to verify the result. It could be referred to in the discussion.
In addition, related to Figure 6, Bertonlini et al., 2019, identified a number of Sox2 responsive enhancers, expressed in the dorsal telencephalon but it is not clear why these are not incorporated into the model. Further justification in the discussion would be helpful. The authors may also consider discussing how Emx2 in their model since they previously showed it was a negative regulator of Sox2 (Mariani et al 2012) and is required for hippocampal development (eg: Pellegrini et al., 1996; Yoshida et al 1997; Zhao et a la 2006).
Regarding the interpretation of the results in Figure 7, previous work by the authors showed that early deletion of Sox2 using a Bf1Cre driver line resulted in severe developmental defects of the ganglionic eminences and therefore GABAergic interneurons. Are the development of GABAergic interneurons affected in the FoxG1Cre cross? It would be preferable to include some analysis of this, or at least a discussion of this issue in the context of the electrophysiology results.
The authors use an eYFP reporter line in Figure 1- supplement. If they have similar data demonstrating Cre activity with the eYFP reporter crossed into the FoxG1CreXSox2flox/flox and Emx1CreXSox2 flox/flox it would be good to add this. It would demonstrate cell autonomous knockout versus non-cell autonomous knockout of Sox2 and may help with the interpretation of Sox2 function.
-
###Reviewer #2:
This study examines the phenotype of early deletion of Sox2 and shows that there is a major dentate phenotype when fl-Sox2 mice are crossed to Foxg1-Cre when compared to Emx1-Cre or Nestin-Cre. This is a novel phenotype, but I don't think the authors have addressed the basis of this phenotype adequately to understand the basis of the phenotype. In addition, I am concerned about a major confounding issue (see below). I believe significant additional studies are needed to establish the specific role of Sox2 here. Below I list the major concerns.
The authors rely on Foxg1-Cre for their main evidence that very early deletion of Sox2 leads to near loss of the dentate. However, it doesn't appear that the authors are aware that Foxg1 het mice have a fairly significant dentate phenotype (see this paper). The Foxg1-Cre line …
###Reviewer #2:
This study examines the phenotype of early deletion of Sox2 and shows that there is a major dentate phenotype when fl-Sox2 mice are crossed to Foxg1-Cre when compared to Emx1-Cre or Nestin-Cre. This is a novel phenotype, but I don't think the authors have addressed the basis of this phenotype adequately to understand the basis of the phenotype. In addition, I am concerned about a major confounding issue (see below). I believe significant additional studies are needed to establish the specific role of Sox2 here. Below I list the major concerns.
The authors rely on Foxg1-Cre for their main evidence that very early deletion of Sox2 leads to near loss of the dentate. However, it doesn't appear that the authors are aware that Foxg1 het mice have a fairly significant dentate phenotype (see this paper). The Foxg1-Cre line generated by Hebert and used by the authors is a knock-in allele that inactivates the endogenous Foxg1 gene. The authors need to address whether the phenotype they observe is actually due to loss of Sox2 alone at E9.5 vs the combined loss of Sox2 and a copy of Foxg1. In particular, could this explain the difference between Emx1-Cre and Foxg1-Cre lines? If this is the explanation for the difference, it isn't clear to me that the story really holds together without bringing in far more complex compound mutant explanations.
The phenotype as described by the authors appears to be most compatible with the published Wnt3a mutant phenotype - perhaps a hypomorphic version makes the most sense or a near phenocopy of the Lef1 mutant. Given this, it appears to me this is really a hem phenotype and is likely explained by the loss of Wnt3a predominantly. Yet the authors don't show direct regulation of Wnt3a by Sox2 - the study would be dramatically enhanced by addressing the mechanism of loss of Wnt3a expression. In addition, examining the expression of Lef1 might reveal the more proximal mechanism of loss of DGC than simply less Wnt3a. This might also be another potential direct target of Sox2 since Lef1 expression is regulated by Wnt signaling but also by other morphogenic signals and could be a Sox2 target.
The authors provide little specific analysis of hippocampal subfield specific markers. Their assumption is that the cells that are in the malformed dentate are granule neurons but they don't use any specific markers of DGC (eg Prox1). Instead they rely on cell position and expression of NeuroD (which is nonspecific). Similarly, it would make sense to examine other markers of mossy cells and CA3, which are also in the same region as DGC and made by adjacent neuroepithelium.
Much of the study relies on the assumption that Nestin-Cre is an efficient deleter in the entire hippocampus yet there is no direct evidence of this. The authors could easily determine when Sox2 expression is lost in the various Cre-deleter lines using antibodies.
I think the electrophysiology section isn't very useful or important. We know that mice with major developmental defects in the DG and hippocampus will have changes in circuit physiology. There is nothing specific about this phenotype, nor does it shed light on the important biology here.
The only two direct targets they find don't seem likely to be important players in the phenotypes they describe, thus, it seems that they don't necessarily address the biology here. The Gli3 phenotypes that have been published are quite distinct from this.
Some of the dentate phenotype is no doubt due to defects in CR cell production or development and this indirect effect has been seen in many other mutants that affect CR cell production (ie a disorganized dentate). It is hard to see how this part of the phenotype, which is likely due to the hem defects (the neuroepithelium that makes the CR cells) is helping us to understand the fundamental aspects of this phenotype.
-
###Reviewer #1:
In the paper by Mercurio et al, the authors examine the role of SOX2 in the development of mouse hippocampal dentate gyrus. Using conditionally mutant SOX2 mice the authors show that early, but not late, deletion of SOX2 leads to developmental impairments of the dentate gyrus. A drawback of their study is that these findings have been reported previously by the group (Favaro et al. 2009; Ferri et al. 2013). In the current study the authors show additional examples of SOX2 target genes, which are dysregulated in the cortical hem upon early SOX2 deletion. However, as no mechanistic insights how this may affect dentate gyrus development are provided, the general novelty of the study is limited.
Comments:
The language of the manuscript needs to be improved.
Using different Cre-drivers the authors aim to delete SOX2 at …
###Reviewer #1:
In the paper by Mercurio et al, the authors examine the role of SOX2 in the development of mouse hippocampal dentate gyrus. Using conditionally mutant SOX2 mice the authors show that early, but not late, deletion of SOX2 leads to developmental impairments of the dentate gyrus. A drawback of their study is that these findings have been reported previously by the group (Favaro et al. 2009; Ferri et al. 2013). In the current study the authors show additional examples of SOX2 target genes, which are dysregulated in the cortical hem upon early SOX2 deletion. However, as no mechanistic insights how this may affect dentate gyrus development are provided, the general novelty of the study is limited.
Comments:
The language of the manuscript needs to be improved.
Using different Cre-drivers the authors aim to delete SOX2 at different developmental stages. What references demonstrate that EMX-Cre first deletes SOX2 after E10.5? I don't find where in Tronche et al. 1999 is it shown that Nes-Cre is deleting after E11.5?
At line 149 the authors state "...remarkably, in the FoxG1-Cre cKO, the DG appears to be almost absent (Figure 2A).". The question is why this finding is remarkable as it already was published in (Ferri et al. 2013).
Line 154 "In the FoxG1-Cre cKO, Reelin expression (marking CRC) is greatly reduced, and a HF is not observed (Figure 2D);...". This statement has no support from Figure 2D.
Some of the images presented in Figure 4 are of such poor quality that they are hard to evaluate.
In Figure 6 the authors show that SOX2 interacts with the promoter region of the Cxcr4 gene and that the SOX2 bound enhancer is active in the developing Zebrafish brain. These data can be removed as they have been published previously in Bertolini et al. 2019.
-
##Preprint Review
This preprint was reviewed using eLife’s Preprint Review service, which provides public peer reviews of manuscripts posted on bioRxiv for the benefit of the authors, readers, potential readers, and others interested in our assessment of the work. This review applies only to version 1 of the manuscript. Joseph G Gleeson (Howard Hughes Medical Institute, The Rockefeller University) served as the Reviewing Editor.
###Summary:
The positive aspects of the paper are the examination of the role of SOX2 in the development of mouse hippocampal dentate gyrus. Using conditionally mutant SOX2 mice the authors show that early, but not late, deletion of SOX2 leads to developmental impairments of the dentate gyrus. There were substantial criticisms of the work, most importantly that the work does not advance the field as much as is …
##Preprint Review
This preprint was reviewed using eLife’s Preprint Review service, which provides public peer reviews of manuscripts posted on bioRxiv for the benefit of the authors, readers, potential readers, and others interested in our assessment of the work. This review applies only to version 1 of the manuscript. Joseph G Gleeson (Howard Hughes Medical Institute, The Rockefeller University) served as the Reviewing Editor.
###Summary:
The positive aspects of the paper are the examination of the role of SOX2 in the development of mouse hippocampal dentate gyrus. Using conditionally mutant SOX2 mice the authors show that early, but not late, deletion of SOX2 leads to developmental impairments of the dentate gyrus. There were substantial criticisms of the work, most importantly that the work does not advance the field as much as is expected for a high-ranking journal, considering prior publications, and that there could be some difficulties interpreting the data as presented.
-
