Dnmt3a knockout in excitatory neurons impairs postnatal synapse maturation and increases the repressive histone modification H3K27me3
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
In this manuscript the authors conditionally knock out the DNA methyltransferase Dnmt3a in developing excitatory cortical neurons to determine the consequences for chromatin regulation, gene expression, and neuron function. As expected they find widespread loss of DNA methylation at CpA dinucleotides but also an increase in histone methylation (H3K27me3) at many similar regions of the genome, which they speculate may be a mechanism of functional compensation. Overall this study offers new insights into the gene regulatory and neuronal cellular functions of an important chromatin regulatory protein.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #3 agreed to share their name with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Two epigenetic pathways of transcriptional repression, DNA methylation and polycomb repressive complex 2 (PRC2), are known to regulate neuronal development and function. However, their respective contributions to brain maturation are unknown. We found that conditional loss of the de novo DNA methyltransferase Dnmt3a in mouse excitatory neurons altered expression of synapse-related genes, stunted synapse maturation, and impaired working memory and social interest. At the genomic level, loss of Dnmt3a abolished postnatal accumulation of CG and non-CG DNA methylation, leaving adult neurons with an unmethylated, fetal-like epigenomic pattern at ~222,000 genomic regions. The PRC2-associated histone modification, H3K27me3, increased at many of these sites. Our data support a dynamic interaction between two fundamental modes of epigenetic repression during postnatal maturation of excitatory neurons, which together confer robustness on neuronal regulation.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
In the manuscript "Dnmt3a knockout impairs synapse maturation and is partly compensated by repressive modification H3K27me3," Li et al. investigate the role of Dnmt3a in the development of mouse cortical neurons by conditionally knocking it out during mid-late gestation and measuring the resulting molecular and phenotypic consequences. The study provides temporal context for Dnmt3a dependent DNA methylation in the development of a specific population of neurons and describes a potentially novel mode of compensatory histone trimethylation at H3K27 at particular genomic loci that lose DNA methylation. The authors first describe phenotypic aberrations induced by Dnmt3a-cko that include altered dendrite/spine morphology and deficits in particular social behaviors without overt morphological …
Author Response
Reviewer #1 (Public Review):
In the manuscript "Dnmt3a knockout impairs synapse maturation and is partly compensated by repressive modification H3K27me3," Li et al. investigate the role of Dnmt3a in the development of mouse cortical neurons by conditionally knocking it out during mid-late gestation and measuring the resulting molecular and phenotypic consequences. The study provides temporal context for Dnmt3a dependent DNA methylation in the development of a specific population of neurons and describes a potentially novel mode of compensatory histone trimethylation at H3K27 at particular genomic loci that lose DNA methylation. The authors first describe phenotypic aberrations induced by Dnmt3a-cko that include altered dendrite/spine morphology and deficits in particular social behaviors without overt morphological alterations in the brain. They then go on to describe the epigenomic landscape underlying their observations.
While the study includes high quality data that are novel, there are a few caveats that need to be addressed. For example, while the manuscript does provide evidence to suggest there may be regions of the genome that are compensated by H3K27me3, the biological basis for this remains unclear, as do the consequences of this compensation. The behavioral data while providing a phenotype for the regulatory role of Dnmt3a in neuronal structure and function are not related in any particular way to the sequencing data. Overall the paper presents chromatin information with a more limited biological context.
We thank the reviewer for appreciating the novelty and quality of our data. While we agree that key questions concerning the biological mechanism and significance of increased H3K27me3 remain, our study sets the stage for such investigation by providing a valid mouse model for excitatory neuron-specific loss of postnatal DNA methylation. Likewise, the behavioral studies we report do not exhaustively define the functional consequences of loss of Dnmt3a in pyramidal neurons, but they provide a foundation by defining the broad cognitive domains (working memory, social interaction) that are impacted. Importantly, our behavioral studies were also important to establish that many key cognitive functions (e.g. learning and memory) are largely preserved despite the massive disruption in epigenetic regulation of a large and critical population of cortical excitatory neurons. These mild behavioral deficits, together with the restricted transcriptional changes, point to some compensatory mechanism being turned on after the loss of Dnmt3a, which we proposed was due to H3K27me3 expansion.
Reviewer #2 (Public Review):
In this study, Li, Pinto-Duarte and colleagues investigate functional and epigenomic effects of loss of DNMT3A in excitatory neurons using a conditional knockout mouse model. The authors characterize behavioral, cell-morphological, and electrophysiological deficits that suggest disruption of synapse function may be major driver of phenotypes in these mice. Through RNAseq analysis of mutant neurons they identify 1720 dysregulated genes, some of which are implicated in dendritic and axonal development and synaptic formation. To understand the epigenetic factors underlying transcriptomic effects, the authors perform methylC-seq. They observe widespread reductions of mCG and mCH in mutant excitatory neurons and detect 141,633 differentially CG methylated regions (DMRs) which exhibit large reductions in mCG. To understand why sets of genes with widespread methylation depletion could be either up- or downregulated, the authors profiled histone modifications. They observe changes in H3K27me3 signal over development and increases in this mark at DMRs upon loss of DNMT3A. They suggest that over-compensation by H3K27me3 repression at genes containing DMRs may drive some of the downregulation of gene expression observed in DNMT3A mutant mice. These results confirm findings from previous publications on loss DNA methylation in DNMT3A conditional mutant mice and identify novel alterations in H3K27me3 that may impact changes in gene expression in these mutants.
Understanding functional outcomes of DNMT3A loss and identifying mechanistic interplay between neuronal DNA methylation and other epigenetic mechanisms is of significant interest to the field. It has been clear that DNMT3A is critical to neuronal development, but cellular characterization such as spine morphology and synapse function has been limited. The analyses presented here provide robust evidence for synaptic alterations upon loss of DNMT3A. The authors' characterization of the differences in H3K27me3 across development and in the DNMT3A cKO underscores the potential importance of this mark when DNA methylation is altered.
We would like to thank the Reviewer for their thoughtful assessment of the significance of our data and findings.
While changes in H3K27me3 are relevant and are likely to be functionally important, the study has some limitations in assessing the magnitude and impact of these changes:
- Only two biological replicates per condition are included in most genomic analysis. This may lead to over-estimates of the changes observed due to sample-specific technical variation in the ChIP and sequencing procedures, particularly given the subtle alterations that are identified.
We appreciate the reviewer’s concern regarding the number of biological replicates, which are critical for ensuring the reproducibility of our findings in independent animals. To reduce variability due to individual differences, the majority of our sequencing data come from tissue samples pooled from two mice. The only exception is MethylC-seq data from P0 mice, where we have 6 control and 2 cKO samples that each came from one individual. This information is now included in the “num_pooled_animals” column in Supplementary Table 1. We have added additional analyses showing the strong consistency of our results across biological replicates for RNA-seq (Figure S8A), MethylC-seq (Figure S10A), and ChIP-seq (Figure S19).
In addition, the current resubmission includes new datasets from two new replicates for both RNA-seq and MethylC-Seq. These data are highly consistent with the previous findings. For example, for the 70 genes which are found to be differentially expressed (FDR < 0.05) in our new batch of RNA-seq data, 53 (75.7%) showed the same direction of expression change (up- or down-regulation) in the previous batch (Fig. R1):
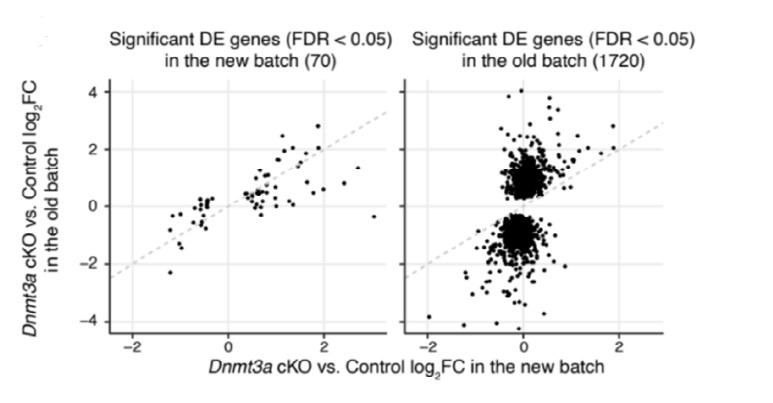
Fig. R1: Scatter to show the consistency of gene expression fold-changes (Dnmt3a cKO vs. control) across the two batches of RNA-seq samples using significant DE genes detected in the new batch (left) and significant DE genes detected in the old batch (right).
- While the compensatory mechanism proposed is feasible in light of the findings presented, evidence definitively supporting H3K27me3 changes as truly compensatory for loss of mCG in DNMT3A conditional knockout neurons is limited. Additional genomic analyses or experimental evidence would be needed to authoritatively make this claim.
We agree that definitively establishing a causal role for the histone methylation changes in compensating for the loss of DNMT3A would require additional experiments, such as manipulation of histone methyltransferases. Such experiments are beyond the scope of this study. We have revised the manuscript to acknowledge this limitation and more clearly state the nature of our conclusions:
"Overall, our results suggest that when DNA methylation is disrupted, H3K27me3 might partially compensate for the loss of mCG and/or mCH and act as an alternative mode of epigenetic repression. Nevertheless, we did not find differential expression in any of the four core components of PRC2 (Ezh2, Suz12, Eed and Rbbp4) in adult Dnmt3a cKO animals. It is possible that the increased H3K27me3 was mediated by transient expression of PRC2 components during development in the cKO. Furthermore, the predictions from BART (Figure 4A) were derived from various cell lines and tissues from the ENCODE project (Davis et al., 2018; ENCODE Project Consortium, 2012), suggesting that the potential PRC2 binding at our DEGs may normally happen in systems other than the brain or pyramidal neurons, or at other time points during development. Additional experiments which directly manipulate components of the PRC2 system are required to further test the potential compensation mechanism."
- The study includes limited analyses assessing how changes in mCH and H3K27ac, two other epigenetic marks shown to be disrupted in DNMT3A models, are integrated with changes in H3K27me3, mCG and gene expression.
We found an increase in H3K27ac, specifically at DMRs which lose mCG in the cKO (shown in Figure 5C). This was an expected finding reflecting the epigenetic activation of enhancer regions that fail to gain DNA methylation.
Regarding mCH, our study was originally motivated by our interest in the role of mCH in neural development, and we were very interested in exploring this question. The complete loss of mCH is indeed a very dramatic effect of the cKO (Figure 3C), and this genome-wide disruption of the normal DNA methylation pattern might have been expected to severely impact neural function. Instead, our data showed relatively limited alterations in neural gene expression, as well as synaptic physiology and social behavior. Thus, although we did analyze the link between mCH and gene expression (e.g. in Figure 3D-E), we found that the loss of mCH could explain only a very small fraction (0.456%) of the differential expression (Supplementary Figure S11D). By contrast, mCG changes occur in a localized fashion specifically in regions that are developmentally regulated and gaining mCG via Dnmt3a during postnatal development. Because we found a clear association between these mCG differences and H3K27me3, we performed a more in-depth analysis on those marks.
Overall, the study has generated valuable datasets that identify cellular phenotypes and suggest a novel disruption of H3K27me3 in DNMT3A conditional knockout mice. However, the conclusions regarding the importance of H3K27me3 in compensation in these mutant mice are quite speculative.
-
Evaluation Summary:
In this manuscript the authors conditionally knock out the DNA methyltransferase Dnmt3a in developing excitatory cortical neurons to determine the consequences for chromatin regulation, gene expression, and neuron function. As expected they find widespread loss of DNA methylation at CpA dinucleotides but also an increase in histone methylation (H3K27me3) at many similar regions of the genome, which they speculate may be a mechanism of functional compensation. Overall this study offers new insights into the gene regulatory and neuronal cellular functions of an important chromatin regulatory protein.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #3 agreed to share their name …
Evaluation Summary:
In this manuscript the authors conditionally knock out the DNA methyltransferase Dnmt3a in developing excitatory cortical neurons to determine the consequences for chromatin regulation, gene expression, and neuron function. As expected they find widespread loss of DNA methylation at CpA dinucleotides but also an increase in histone methylation (H3K27me3) at many similar regions of the genome, which they speculate may be a mechanism of functional compensation. Overall this study offers new insights into the gene regulatory and neuronal cellular functions of an important chromatin regulatory protein.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #3 agreed to share their name with the authors.)
-
Reviewer #1 (Public Review):
In the manuscript "Dnmt3a knockout impairs synapse maturation and is partly compensated by repressive modification H3K27me3," Li et al. investigate the role of Dnmt3a in the development of mouse cortical neurons by conditionally knocking it out during mid-late gestation and measuring the resulting molecular and phenotypic consequences. The study provides temporal context for Dnmt3a dependent DNA methylation in the development of a specific population of neurons and describes a potentially novel mode of compensatory histone trimethylation at H3K27 at particular genomic loci that lose DNA methylation. The authors first describe phenotypic aberrations induced by Dnmt3a-cko that include altered dendrite/spine morphology and deficits in particular social behaviors without overt morphological alterations in the …
Reviewer #1 (Public Review):
In the manuscript "Dnmt3a knockout impairs synapse maturation and is partly compensated by repressive modification H3K27me3," Li et al. investigate the role of Dnmt3a in the development of mouse cortical neurons by conditionally knocking it out during mid-late gestation and measuring the resulting molecular and phenotypic consequences. The study provides temporal context for Dnmt3a dependent DNA methylation in the development of a specific population of neurons and describes a potentially novel mode of compensatory histone trimethylation at H3K27 at particular genomic loci that lose DNA methylation. The authors first describe phenotypic aberrations induced by Dnmt3a-cko that include altered dendrite/spine morphology and deficits in particular social behaviors without overt morphological alterations in the brain. They then go on to describe the epigenomic landscape underlying their observations.
While the study includes high quality data that are novel, there are a few caveats that need to be addressed. For example, while the manuscript does provide evidence to suggest there may be regions of the genome that are compensated by H3K27me3, the biological basis for this remains unclear, as do the consequences of this compensation. The behavioral data while providing a phenotype for the regulatory role of Dnmt3a in neuronal structure and function are not related in any particular way to the sequencing data. Overall the paper presents chromatin information with a more limited biological context.
-
Reviewer #2 (Public Review):
In this study, Li, Pinto-Duarte and colleagues investigate functional and epigenomic effects of loss of DNMT3A in excitatory neurons using a conditional knockout mouse model. The authors characterize behavioral, cell-morphological, and electrophysiological deficits that suggest disruption of synapse function may be major driver of phenotypes in these mice. Through RNA-seq analysis of mutant neurons they identify 1720 dysregulated genes, some of which are implicated in dendritic and axonal development and synaptic formation. To understand the epigenetic factors underlying transcriptomic effects, the authors perform methylC-seq. They observe widespread reductions of mCG and mCH in mutant excitatory neurons and detect 141,633 differentially CG methylated regions (DMRs) which exhibit large reductions in mCG. To …
Reviewer #2 (Public Review):
In this study, Li, Pinto-Duarte and colleagues investigate functional and epigenomic effects of loss of DNMT3A in excitatory neurons using a conditional knockout mouse model. The authors characterize behavioral, cell-morphological, and electrophysiological deficits that suggest disruption of synapse function may be major driver of phenotypes in these mice. Through RNA-seq analysis of mutant neurons they identify 1720 dysregulated genes, some of which are implicated in dendritic and axonal development and synaptic formation. To understand the epigenetic factors underlying transcriptomic effects, the authors perform methylC-seq. They observe widespread reductions of mCG and mCH in mutant excitatory neurons and detect 141,633 differentially CG methylated regions (DMRs) which exhibit large reductions in mCG. To understand why sets of genes with widespread methylation depletion could be either up- or down-regulated, the authors profiled histone modifications. They observe changes in H3K27me3 signal over development and increases in this mark at DMRs upon loss of DNMT3A. They suggest that over-compensation by H3K27me3 repression at genes containing DMRs may drive some of the downregulation of gene expression observed in DNMT3A mutant mice. These results confirm findings from previous publications on loss DNA methylation in DNMT3A conditional mutant mice and identify novel alterations in H3K27me3 that may impact changes in gene expression in these mutants.
Understanding functional outcomes of DNMT3A loss and identifying mechanistic interplay between neuronal DNA methylation and other epigenetic mechanisms is of significant interest to the field. It has been clear that DNMT3A is critical to neuronal development, but cellular characterization such as spine morphology and synapse function has been limited. The analyses presented here provide robust evidence for synaptic alterations upon loss of DNMT3A. The authors' characterization of the differences in H3K27me3 across development and in the DNMT3A cKO underscores the potential importance of this mark when DNA methylation is altered. While changes in H3K27me3 are relevant and are likely to be functionally important, the study has some limitations in assessing the magnitude and impact of these changes:
1. Only two biological replicates per condition are included in most genomic analysis. This may lead to over-estimates of the changes observed due to sample-specific technical variation in the ChIP and sequencing procedures, particularly given the subtle alterations that are identified.
2. While the compensatory mechanism proposed is feasible in light of the findings presented, evidence definitively supporting H3K27me3 changes as truly compensatory for loss of mCG in DNMT3A conditional knockout neurons is limited. Additional genomic analyses or experimental evidence would be needed to authoritatively make this claim.
3. The study includes limited analyses assessing how changes in mCH and H3K27ac, two other epigenetic marks shown to be disrupted in DNMT3A models, are integrated with changes in H3K27me3, mCG and gene expression.
Overall, the study has generated valuable datasets that identify cellular phenotypes and suggest a novel disruption of H3K27me3 in DNMT3A conditional knockout mice. However, the conclusions regarding the importance of H3K27me3 in compensation in these mutant mice are quite speculative.
-
Reviewer #3 (Public Review):
In order to understand the regulatory mechanisms whereby DNMT3A controls neuronal maturation, the authors generate and analyze an extensive set of transcriptomic and epigenomic datasets generated in WT and DNMT3A KO neurons. However, in my opinion, these analyses provide largely correlative rather than causative observations, in which the direct and secondary effects of DNMT3A and DNA methylation on gene expression can not be distinguished from each other. This is well illustrated by the proposed mechanism whereby the accumulation of H3K27me3 in DNMT3A KO neurons can compensate the loss of DNA methylation in order to maintain the repression of certain genes and thus confer robustness to neuronal maturation. However, such compensatory mechanism is only supported by the increased levels of H3K27me3 around the …
Reviewer #3 (Public Review):
In order to understand the regulatory mechanisms whereby DNMT3A controls neuronal maturation, the authors generate and analyze an extensive set of transcriptomic and epigenomic datasets generated in WT and DNMT3A KO neurons. However, in my opinion, these analyses provide largely correlative rather than causative observations, in which the direct and secondary effects of DNMT3A and DNA methylation on gene expression can not be distinguished from each other. This is well illustrated by the proposed mechanism whereby the accumulation of H3K27me3 in DNMT3A KO neurons can compensate the loss of DNA methylation in order to maintain the repression of certain genes and thus confer robustness to neuronal maturation. However, such compensatory mechanism is only supported by the increased levels of H3K27me3 around the promoter regions of some genes, but whether this increased levels of H3K27me3 have any functional impact on gene expression and/or in neuronal function has not been addressed by the authors. Similarly, the authors uncovered a large number of genomic regions that lose DNA methylation in DNMT3A KO neurons (differentially methylated regions (DMRs)) and that frequently overlap with putative enhancers. Based on these observations, the authors suggest that DNMT3A is essential for the methylation and subsequent repression of neuronal enhancers that are active during prenatal brain development. However, these suggestions are not fully supported by the data, as the authors do not provide direct evidences of whether the loss of DNA methylation in enhancers has any impact on their activity (e.g. H3K27ac or eRNA levels) or function (e.g. changes in expression of target genes).
More generally, recent work in the epigenetics field suggests that the function of certain epigenetic regulators might not be directly linked to their associated epigenetic marks (e.g. MLL3/4 and H3K4me1). To conclusively assess whether the functional relevance of DNMT3A or PRC2 could be attributed to their enzymatic activities and associated epigenetic marks, the generation of catalytic mutants would be ideal. Generating these catalytic mutants in a conditional manner can be technically challenging. Therefore, in the absence of such mutants, strong claims regarding the functional relevance of epigenetic marks should be avoided.
-
