Deep-sequence phylogenetics to quantify patterns of HIV transmission in the context of a universal testing and treatment trial – BCPP/Ya Tsie trial
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
One hypothesis to explain recent worse-than-expected outcomes of universal test-and-treat HIV prevention trials is population mobility. The authors show phylogenetically that mobility could play a role in transmission events in a large trial in Botswana. This study is of public health interest, has a large sample size for a phylogenetic study in this setting, and overall precise analysis. A few methodological clarifications are still needed to ensure that the data supports the study's claims.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 agreed to share their name with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Mathematical models predict that community-wide access to HIV testing-and-treatment can rapidly and substantially reduce new HIV infections. Yet several large universal test-and-treat HIV prevention trials in high-prevalence epidemics demonstrated variable reduction in population-level incidence.
Methods:
To elucidate patterns of HIV spread in universal test-and-treat trials, we quantified the contribution of geographic-location, gender, age, and randomized-HIV-intervention to HIV transmissions in the 30-community Ya Tsie trial in Botswana. We sequenced HIV viral whole genomes from 5114 trial participants among the 30 trial communities.
Results:
Deep-sequence phylogenetic analysis revealed that most inferred HIV transmissions within the trial occurred within the same or between neighboring communities, and between similarly aged partners. Transmissions into intervention communities from control communities were more common than the reverse post-baseline (30% [12.2 – 56.7] vs. 3% [0.1 – 27.3]) than at baseline (7% [1.5 – 25.3] vs. 5% [0.9 – 22.9]) compatible with a benefit from treatment-as-prevention.
Conclusions:
Our findings suggest that population mobility patterns are fundamental to HIV transmission dynamics and to the impact of HIV control strategies.
Funding:
This study was supported by the National Institute of General Medical Sciences (U54GM088558), the Fogarty International Center (FIC) of the U.S. National Institutes of Health (D43 TW009610), and the President’s Emergency Plan for AIDS Relief through the Centers for Disease Control and Prevention (CDC) (Cooperative agreements U01 GH000447 and U2G GH001911).
Article activity feed
-

-

Author Response:
Reviewer #1:
Weaknesses:
For me, most of the weaknesses of this manuscript are related to the cluster detection:
- There is no consensus on the definition of transmission clusters in the field. However, the rational of taking the union (rather than the intersection) of two different methods (HIV-TRACE and cluster picker) did not become clear to me.
- HIV-TRACE defines clusters based on pairwise genetic distances and cluster picker identifies clusters using pairwise genetic distance with the guidance of a phylogenetic tree (and node support / bootstrap values). Given the underlying sample size and that the phylogeny was constructed already, the rationale for the purely distance related criterion of HIV-TRACE did not become clear.
We thank the reviewer for their comments and are happy to provide additional results …
Author Response:
Reviewer #1:
Weaknesses:
For me, most of the weaknesses of this manuscript are related to the cluster detection:
- There is no consensus on the definition of transmission clusters in the field. However, the rational of taking the union (rather than the intersection) of two different methods (HIV-TRACE and cluster picker) did not become clear to me.
- HIV-TRACE defines clusters based on pairwise genetic distances and cluster picker identifies clusters using pairwise genetic distance with the guidance of a phylogenetic tree (and node support / bootstrap values). Given the underlying sample size and that the phylogeny was constructed already, the rationale for the purely distance related criterion of HIV-TRACE did not become clear.
We thank the reviewer for their comments and are happy to provide additional results that motivate our decision to use the union of clusters detected with HIV-TRACE and Cluster Picker to estimate HIV transmissions within and between demographic sub-groups in the Botswana - Ya Tsie trial population. The primary motivation was that a filtering step was required to save time and computational resources from evaluating sequences that were too distantly related, before applying the “gold standard” of Phyloscanner to detect directed (when possible) transmission pairs. Accordingly, clustering algorithms plus a distance threshold helped to achieve this filtering. Because we shared what we take to be the reviewers’ concerns about either of the algorithms alone, we sought to maximize the number of transmission pairs that could be identified between participants in the Botswana – Ya Tsie trial with Phyloscanner by using the union of clusters detected with HIV-TRACE and Cluster Picker. This also served as a sensitivity analysis that allowed us to evaluate the extent to which the clustering patterns observed were specific to a single algorithm.
Furthermore, a previous study done by Rose and colleagues (PMID: 27824249) to compare the number and size of clusters identified with HIV-TRACE and Cluster Picker clustering algorithms revealed that HIV-TRACE generally identified larger but fewer clusters, compared with clusters identified with Cluster Picker that were typically more numerous and mostly small 2-person clusters (Please see Figure 3B below extracted from Rose and colleagues (PMID: 27824249)). This suggested that HIV-TRACE would be helpful in detecting potentially larger transmission chains and Cluster Picker would be valuable in revealing potential transmission events between pairs of individuals.
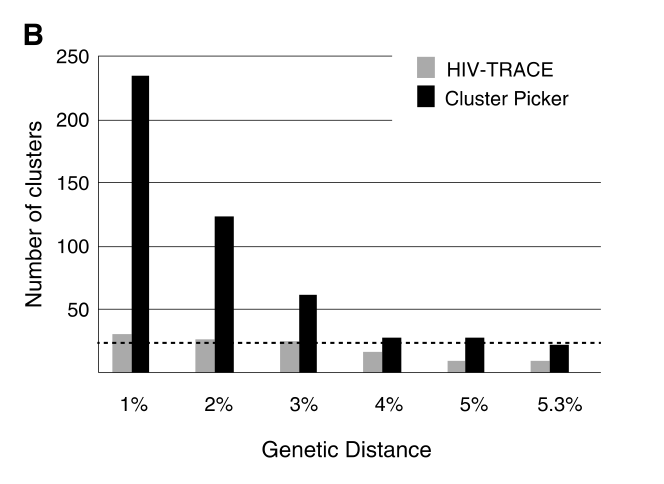
Of the 236 genetic clusters detected with the two algorithms, we identified 19 full or partial clusters (including 41 sequences) that included members that were only detected with HIV- TRACE and 122 full or partial clusters (including 242 sequences) that were unique to Cluster Picker. Moreover, of the 82 directed male-female transmission pairs inferred from the sample, (n = 5) were from genetic clusters that were unique to HIV-TRACE compared with (n = 27) that were from clusters unique to Cluster Picker. Of the five transmission events unique to HIV- TRACE clusters, three occurred in intervention communities originating from control communities. By contrast, four of the twenty-seven transmission events unique to Cluster Picker clusters occurred in intervention communities from control communities.
In summary, estimates of HIV transmissions in the trial population based on the full overlap of clusters detected with HIV-TRACE and Cluster Picker would have excluded 32 of the 82 male- female pairs used for the primary analysis.
- For a phylogeny of this size it is feasible to calculate real bootstrap values instead of using (in my experience more liberal) Shimodaira-Hasegawa support values.
We value the reviewer suggestion and agree that real bootstrap values could be ideal. However, the likely benefit of computing the suggested bootstrap values and thereafter repeating the entire analysis inferring transmission pairs with Phyloscanner and estimating transmission flows would be minimal. As noted above, liberality in a filtering step is a virtue (avoiding filtering out pairs of interest) as long as it does not lead to unfeasibly large computational burden, as this did not.
- In Supplementary Note 2.5 it is described how the linkage and direction of transmission score threshold of 57% was chosen. However, the finding that almost half of the accordingly selected probable source-recipient pairs were same-sex and had to be excluded from the analysis questions the reliability of the threshold.
We apologize for the insufficient clarity in our description and would like the reviewer to kindly note that the threshold in of itself is insufficient to distinguish between Female-Female pairs separated by a single Male intermediate, but rather by design can distinguish between direct Male-Female pairs and Male-Female pairs separated by several intermediates. Once again, the threshold was meant to be a filter that would allow us to run Phyloscanner on a feasible number of sequences, thus appropriately should let through some pairs that are rejected by later steps in the pipeline. Also, kindly note that all previous Supplementary Notes are now presented in the methods section in line with the reviewer’s suggestions.
-
Evaluation Summary:
One hypothesis to explain recent worse-than-expected outcomes of universal test-and-treat HIV prevention trials is population mobility. The authors show phylogenetically that mobility could play a role in transmission events in a large trial in Botswana. This study is of public health interest, has a large sample size for a phylogenetic study in this setting, and overall precise analysis. A few methodological clarifications are still needed to ensure that the data supports the study's claims.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 agreed to share their name with the authors.)
-
Reviewer #1 (Public Review):
The study aims at phylogenetically identifying determinants of HIV transmission dynamics in an HIV prevention trial in Botswana. The study found that most HIV transmissions occurred between similarly aged partners within the same trial community or between trial communities in close proximity. Interestingly, there was a greater flow of HIV transmissions into intervention communities from control communities than vice versa - which could potentially explain worth-than-expected outcomes of such prevention trials.
Strengths:
1. This research question is of public health relevance.
2. The sample size was large for a phylogenetic study in this setting. This is due to the prospective planning of the study, which enabled sequencing proviral DNA of the large fraction of participants with suppressed viral load.
3. …Reviewer #1 (Public Review):
The study aims at phylogenetically identifying determinants of HIV transmission dynamics in an HIV prevention trial in Botswana. The study found that most HIV transmissions occurred between similarly aged partners within the same trial community or between trial communities in close proximity. Interestingly, there was a greater flow of HIV transmissions into intervention communities from control communities than vice versa - which could potentially explain worth-than-expected outcomes of such prevention trials.
Strengths:
1. This research question is of public health relevance.
2. The sample size was large for a phylogenetic study in this setting. This is due to the prospective planning of the study, which enabled sequencing proviral DNA of the large fraction of participants with suppressed viral load.
3. Overall precise analysis: Efforts to adjust the analysis for sampling density, weight the mean age gap etc.
4. A large part of the limitations of this study was discussed.
5. The figures are an added value and nicely made.
6. A large amount of supplementary information is available for the interested reader.
7. An R package with all code will be made available.Weaknesses:
For me, most of the weaknesses of this manuscript are related to the cluster detection:
8. There is no consensus on the definition of transmission clusters in the field. However, the rational of taking the union (rather than the intersection) of two different methods (HIV-TRACE and cluster picker) did not become clear to me.
9. HIV-TRACE defines clusters based on pairwise genetic distances and cluster picker identifies clusters using pairwise genetic distance with the guidance of a phylogenetic tree (and node support / bootstrap values). Given the underlying sample size and that the phylogeny was constructed already, the rationale for the purely distance related criterion of HIV-TRACE did not become clear.
10. For a phylogeny of this size it is feasible to calculate real bootstrap values instead of using (in my experience more liberal) Shimodaira-Hasegawa support values.
11. In Supplementary Note 2.5 it is described how the linkage and direction of transmission score threshold of 57% was chosen. However, the finding that almost half of the accordingly selected probable source-recipient pairs were same-sex and had to be excluded from the analysis questions the reliability of the threshold.Conclusions are justified by the data:
If the authors can justify their cluster definitions, I believe that their conclusions are justified.
Discussion:
This study might help to understand results of past HIV prevention trials and impact the design of future HIV prevention trials. Further, it provides methodological tools for HIV phylogenetic studies in similar settings.
-
Reviewer #2 (Public Review):
Using a case control trial design, Magosi et al., describe an impressively large study that makes use of deep viral sequencing to investigate the contributions location, gender, age and ready access to HIV care have in the transmission of HIV between people in Botswana. They identified that most transmission occurs within close geographic proximity, that increased access to HIV care can significantly reduce transmission, and that men and women contributed similarly to the spread of infection in phylogenetically linked pairs. They also introduce the program 'bumblebee' which provides methods for estimating transmission flows between populations. This study is highly relevant for the interpretation of other universal test-and-treat HIV prevention trials and in the design of HIV prevention programmes more …
Reviewer #2 (Public Review):
Using a case control trial design, Magosi et al., describe an impressively large study that makes use of deep viral sequencing to investigate the contributions location, gender, age and ready access to HIV care have in the transmission of HIV between people in Botswana. They identified that most transmission occurs within close geographic proximity, that increased access to HIV care can significantly reduce transmission, and that men and women contributed similarly to the spread of infection in phylogenetically linked pairs. They also introduce the program 'bumblebee' which provides methods for estimating transmission flows between populations. This study is highly relevant for the interpretation of other universal test-and-treat HIV prevention trials and in the design of HIV prevention programmes more broadly.
Overall, the data were well analysed. However, some methods require further explanation and motivation.
-
