Magnesium isoglycyrrhizinate alleviates alcohol-associated liver disease through targeting HSD11B1
Curation statements for this article:-
Curated by eLife

eLife Assessment
This important study reports characterisation of hepatocyte molecular pathways affected by a glycyrrhizin derivative in both in vivo and in vitro mouse models of alcohol-associated liver disease. The authors show convincing evidence indicating that IPP delta isomerase 1 (Idi1) is an intermediate in these pharmacological effects, via the binding of the glycyrrhizin derivative to an upstream regulator of Idi1, HSD11B1, although some more quantitative analyses and better organisation of data would strengthen the study. The findings would be of interest to immunologists and pharmacologists interested in liver inflammation and its amelioration.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
While magnesium isoglycyrrhizinate (MgIG) is a clinically approved therapy for alcohol-associated liver disease (ALD), its precise molecular targets and mechanisms remain uncharacterized. This study aimed to define MgIG’s hepatoprotective actions in chronic-binge ALD mouse models and ethanol/palmitic acid-exposed AML-12 hepatocytes. Through an integrated strategy encompassing RNA sequencing, molecular docking, and microscale thermophoresis, we discovered that MgIG directly binds to hydroxysteroid 11-beta dehydrogenase 1 (HSD11B1) at residue 187, a finding corroborated by molecular dynamics simulations. In vivo, MgIG markedly attenuated alcohol-induced liver injury, evidenced by ameliorated histological damage, reduced hepatic steatosis, and normalized liver-to-body weight ratios. In vitro, it effectively reduced lipid accumulation, inflammation, and apoptosis. Mechanistically, RNA sequencing identified isopentenyl diphosphate delta isomerase 1 (IDI1) as a key downstream effector. Hepatocyte-specific genetic manipulations confirmed that MgIG modulates the SREBP2-IDI1 axis, thereby suppressing lipogenesis, inflammatory responses, and apoptotic pathways. We reveal HSD11B1 as a novel direct molecular target of MgIG and elucidate its therapeutic mechanism through the HSD11B1-SREBP2-IDI1 signaling axis, which profoundly impacts ALD pathogenesis. These findings not only validate MgIG’s clinical utility but also highlight a promising new therapeutic target for ALD.
Article activity feed
-

eLife Assessment
This important study reports characterisation of hepatocyte molecular pathways affected by a glycyrrhizin derivative in both in vivo and in vitro mouse models of alcohol-associated liver disease. The authors show convincing evidence indicating that IPP delta isomerase 1 (Idi1) is an intermediate in these pharmacological effects, via the binding of the glycyrrhizin derivative to an upstream regulator of Idi1, HSD11B1, although some more quantitative analyses and better organisation of data would strengthen the study. The findings would be of interest to immunologists and pharmacologists interested in liver inflammation and its amelioration.
-
Reviewer #1 (Public review):
Summary:
In this article by Xiao et al., the authors aimed to identify the precise targets by which magnesium isoglycyrrhizinate (MgIG) functions to improve liver injury in response to ethanol treatment. The authors found through a series of in vivo and molecular approaches that MgIG treatment attenuates alcohol-induced liver injury through a potential SREBP2-IdI1 axis. This manuscript adds to a previous set of literature showing MgIG improves liver function across a variety of etiologies, and also provides mechanistic insight into its mechanism of action.
Strengths:
(1) The authors use a combination of approaches from both in-vivo mouse models to in-vitro approaches with AML12 hepatocytes to support the notion that MgIG does improve liver function in response to ethanol treatment.
(2) The authors use both …
Reviewer #1 (Public review):
Summary:
In this article by Xiao et al., the authors aimed to identify the precise targets by which magnesium isoglycyrrhizinate (MgIG) functions to improve liver injury in response to ethanol treatment. The authors found through a series of in vivo and molecular approaches that MgIG treatment attenuates alcohol-induced liver injury through a potential SREBP2-IdI1 axis. This manuscript adds to a previous set of literature showing MgIG improves liver function across a variety of etiologies, and also provides mechanistic insight into its mechanism of action.
Strengths:
(1) The authors use a combination of approaches from both in-vivo mouse models to in-vitro approaches with AML12 hepatocytes to support the notion that MgIG does improve liver function in response to ethanol treatment.
(2) The authors use both knockdown and overexpression approaches, in vivo and in vitro, to support most of the claims provided.
(3) Identification of HSD11B1 as the protein target of MgIG, as well as confirmation of direct protein-protein interactions between HSD11B1/SREBP2/IDI1, is novel.
Weaknesses:
Major weaknesses can be classified into 3 groups:
(1) The results do not support some claims made.
(2) Qualitative analyses of some of the lipid measures, as opposed to more quantitative analyses.
(3) There are no appropriate readouts of Srebp2 translocation and/or activity.
More specific comments:
(1) A few of the claims made are not supported by the references provided. For instance, line 76 states MgIG has hepatoprotective properties and improved liver function, but the reference provided is in the context of myocardial fibrosis.
(2) MgIG is clinically used for the treatment of liver inflammatory disease in China and Japan. In the first line of the abstract, the authors noted that MgIG is clinically approved for ALD. In which countries is MgIG approved for clinical utility in this space?
(3) Serum TGs are not an indicator of liver function. Alterations in serum TGs can occur despite changes in liver function.
(4) There are discrepancies in the results section and the figure legends. For example, line 302 states Idil is upregulated in alcohol fed mice relative to the control group. The figure legend states that the comparison for Figure 2A is that of ALD+MgIG and ALD only.
(5) Oil Red O staining provided does not appear to be consistent with the quantification in Figure 1D. ORO is nonspecific and can be highly subjective. The representative image in Figure 1C appears to have a much greater than 30% ORO (+) area.
(6) The connection between Idil expression in response to EtOH/PA treatment in AML12 cells with viability and apoptosis isn't entirely clear. MgIG treatment completely reduces Idi1 expression in response to EtOH/PA, but only moderate changes, at best, are observed in viability and apoptosis. This suggests the primary mechanism related to MgIG treatment may not be via Idi1.
(7) The nile red stained images also do not appear representative with its quantification. Several claims about more or less lipid accumulation across these studies are not supported by clear differences in nile red.
(8) The authors make a comment that Hsd11b1 expression is quite low in AML12 cells. So why did the authors choose to knockdown Hsd11b1 in this model?
(9) Line 380 - the claim that MGIG weakens the interaction between HSD11b1 and SREBP2 cannot be made solely based on one Western blot.
(10) It's not clear what the numbers represent on top of the Western blots. Are these averages over the course of three independent experiments?
(11) The claim in line 382 that knockdown of Hsd11b1 resulted in accumulation of pSREBP2 is not supported by the data provided in Figure 6D.
(12) None of the images provided in Figure 6E support the claims stated in the results. Activation of SREBP2 leads to nuclear translocation and subsequent induction of genes involved in cholesterol biosynthesis and uptake. Manipulation of Hsd11b1 via OE or KD does not show any nuclear localization with DAPI.
(13) The entire manuscript is focused on this axis of MgIG-Hsd11b1-Srebp2, but no Srebp2 transcriptional targets are ever measured.
(14) Acc1 and Scd1 are Srebp1 targets, not Srebp2.
(15) A major weakness of this manuscript is the lack of studies providing quantitative assessments of Srebp2 activation and true liver lipid measurements.
-
Reviewer #2 (Public review):
Summary:
In this manuscript, the authors investigated magnesium isoglycyrrhizinate (MgIG)'s hepatoprotective actions in chronic-binge alcohol-associated liver disease (ALD) mouse models and ethanol/palmitic acid-challenged AML-12 hepatocytes. They found that MgIG markedly attenuated alcohol-induced liver injury, evidenced by ameliorated histological damage, reduced hepatic steatosis, and normalized liver-to-body weight ratios. RNA sequencing identified isopentenyl diphosphate delta isomerase 1 (IDI1) as a key downstream effector. Hepatocyte-specific genetic manipulations confirmed that MgIG modulates the SREBP2-IDI1 axis. The mechanistic studies suggested that MgIG could directly target HSD11B1 and modulate the HSD11B1-SREBP2-IDI1 axis to attenuate ALD. This manuscript is of interest to the research field of …
Reviewer #2 (Public review):
Summary:
In this manuscript, the authors investigated magnesium isoglycyrrhizinate (MgIG)'s hepatoprotective actions in chronic-binge alcohol-associated liver disease (ALD) mouse models and ethanol/palmitic acid-challenged AML-12 hepatocytes. They found that MgIG markedly attenuated alcohol-induced liver injury, evidenced by ameliorated histological damage, reduced hepatic steatosis, and normalized liver-to-body weight ratios. RNA sequencing identified isopentenyl diphosphate delta isomerase 1 (IDI1) as a key downstream effector. Hepatocyte-specific genetic manipulations confirmed that MgIG modulates the SREBP2-IDI1 axis. The mechanistic studies suggested that MgIG could directly target HSD11B1 and modulate the HSD11B1-SREBP2-IDI1 axis to attenuate ALD. This manuscript is of interest to the research field of ALD.
Strengths:
The authors have performed both in vivo and in vitro studies to demonstrate the action of magnesium isoglycyrrhizinate on hepatocytes and an animal model of alcohol-associated liver disease.
Weaknesses:
The data were not well-organised, and the paper needs proofreading again, with a focus on the use of scientific language throughout.
Here are several comments:
(1) In Supplemental Figure 1A, all the treatment arms (A-control, MgIG-25 mg/kg, MgIG-50 mg/kg) showed body weight loss compared to the untreated controls. However, Figure 1E showed body weight gain in the treatment arms (A-control and MgIG-25 mg/kg), why? In Supplemental Figure 1A, the mice with MgIG (25 mg/kg) showed the lowest body weight, compared to either A-control or MgIG (50 mg/kg) treatment. Can the authors explain why MgIG (25 mg/kg) causes bodyweight loss more than MgIG (50 mg/kg)? What about the other parameters (ALT, ALS, NAS, etc.) for the mice with MgIG (50 mg/kg)?
(2) IL-6 is a key pro-inflammatory cytokine significantly involved in ALD, acting as a marker of ALD severity. Can the authors explain why MgIG 1.0 mg/ml shows higher IL-6 gene expression than MgIG (0.1-0.5 mg/ml)? Same question for the mRNA levels of lipid metabolic enzymes Acc1 and Scd1.
(3) For the qPCR results of Hsd11b1 knockdown (siRNA) and Hsd11b1 overexpression (plasmid) in AML-12 cells (Figure 5B), what is the description for the gene expression level (Y axis)? Fold changes versus GAPDH? Hsd11b1 overexpression showed non-efficiency (20-23, units on Y axis), even lower than the Hsd11b1 knockdown (above 50, units on Y axis). The authors need to explain this. For the plasmid-based Hsd11b1 overexpression, why does the scramble control inhibit Hsd11b1 gene expression (less than 2, units on the Y axis)? Again, this needs to be explained.
-
Author response:
Thank you for your letter and for the constructive feedback from the reviewers on our manuscript (eLife-RP-RA-2025-109174). We appreciate the time and expertise you and the reviewers have dedicated to improving our work.
We have carefully considered all comments and have developed a comprehensive revision plan. To address the primary concerns, we will conduct several new experiments designed to provide robust support for our key conclusions. Other points will be addressed through textual revisions, including the addition of existing ADMET data and an expanded discussion section.
We are confident that these revisions will fully satisfy the reviewers' concerns and significantly strengthen the manuscript. Our detailed experimental plan and point-by-point responses are provided below.
(1) Addressing "Qualitative analyses …
Author response:
Thank you for your letter and for the constructive feedback from the reviewers on our manuscript (eLife-RP-RA-2025-109174). We appreciate the time and expertise you and the reviewers have dedicated to improving our work.
We have carefully considered all comments and have developed a comprehensive revision plan. To address the primary concerns, we will conduct several new experiments designed to provide robust support for our key conclusions. Other points will be addressed through textual revisions, including the addition of existing ADMET data and an expanded discussion section.
We are confident that these revisions will fully satisfy the reviewers' concerns and significantly strengthen the manuscript. Our detailed experimental plan and point-by-point responses are provided below.
(1) Addressing "Qualitative analyses of some of the lipid measures, as opposed to more quantitative analyses"
Supplementary experiments and analyses
We will add the assessment of hepatic triglyceride and total cholesterol levels in liver tissues from control, experimental, and drug-treated mice, thereby providing further quantitative validation.
(2) Addressing "SREBP2"
Supplementary experiments and analyses
We will include a luciferase assay to determine whether alcohol plus PA induces SREBP2 activation in AML-12 cells.
As suggested, we will assess the expression levels of SREBP2 downstream target genes (Hmgcr, Hmgcs, Ldlr, and Lcn2) in both in vitro and in vivo models.
(3) Timeline and process arrangement of supplementary experiments
To comprehensively address these issues, we plan to purchase the following required reagents and have formulated the following experimental plan:
Author response table 1.
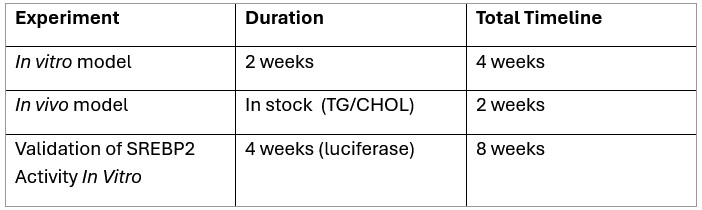
Given the time required for reagent acquisition and the execution of these in vitro and in vivo experiments, we kindly request an extension of the revision deadline by 8 weeks. This will ensure the comprehensive and high-quality completion of all necessary studies.
We will fully commit to delivering a thoroughly revised manuscript that robustly addresses all reviewer comments and aligns with the high standards of eLife. We greatly appreciate your guidance and flexibility.
-

-
-
