Differential interfacial tension between oncogenic and wild-type populations forms the mechanical basis of tissue-specific oncogenesis in epithelia
Curation statements for this article:-
Curated by eLife

eLife Assessment
This important study reports that an oncogenic population in an epithelium can either be repressed or spread, depending on the tissues. This is explained based on the differential interfacial tension hypothesis, and supported by pharmacological perturbations and numerical simulations using the vertex model. The study conveys a key message, but, as it stands, the strength of evidence is incomplete, and a more detailed analysis of the mechanistic origin of the different tensions and better comparison between experiments and simulations would strongly strengthen the message.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Abstract
Why does the same oncogenic mutation drive tumor formation in some tissues but not in others? While cancer driver mutations are well-documented, their tissue-specific effects remain largely attributed to genetic factors, leaving the biophysical aspects underexplored. Here, we demonstrate that mechanical interactions, specifically interfacial tension between newly transformed and wildtype epithelial cells are critical in determining survival and growth of HRasV12 oncogenic mutants in human mammary and bronchial epithelia, leading to contrasting outcomes in the two tissues. In mammary epithelium, isolated oncogenic cells are extruded-a typical mechanism of defense against cancer in epithelia-while oncogenic groups become spatially confined in a kinetically arrested, jammed state, marked by an actomyosin belt at the interface. In contrast, bronchial epithelium permits persistent spreading of the same oncogenic cells, which form long protrusions regardless of colony size. Furthermore, oncogenic clusters in these two tissues exhibit distinct biophysical properties, including variations in cell shapes, intracellular pressure, cell-cell tension, and cellular motility. Using a cell shape-tension coupled bi-disperse vertex model, we reveal that differences in interfacial tension at mutant–wild-type boundaries dictate whether oncogenic cells are eliminated, restrained, or expanded and that modulating interfacial tension alters mutant cell fate within the epithelium. Together, our findings uncover a mechanical basis for tissue-specific oncogenesis by highlighting how differential cellular mechanics at the oncogenic– host cell interface regulate tumor initiation and progression.
Article activity feed
-

eLife Assessment
This important study reports that an oncogenic population in an epithelium can either be repressed or spread, depending on the tissues. This is explained based on the differential interfacial tension hypothesis, and supported by pharmacological perturbations and numerical simulations using the vertex model. The study conveys a key message, but, as it stands, the strength of evidence is incomplete, and a more detailed analysis of the mechanistic origin of the different tensions and better comparison between experiments and simulations would strongly strengthen the message.
-
Reviewer #1 (Public review):
Summary:
The behaviour of cells expressing constitutively active HRas is examined in mosaic monolayers, both in MCF10a breast epithelial and Beas2b bronchial epithelial cell lines, mimicking the potential initial phase of development of carcinoma. Single HRas-positive cells are excluded from MCF10a but not Beas2b monolayers. Most interestingly, however, when in groups, these cells are not excluded, but rather sharply segregated within a MCF10a monolayer. In contrast, they freely mix with wt Beas2b cells. Biophysical analysis identifies high tension at heterotypic interfaces between HRas and wild-type cells as the likely reason for segregation of MCF10a cells. The hypothesis is supported experimentally, as myosin inhibition abolishes segregation. The probable reason for the lack of segregation in the …
Reviewer #1 (Public review):
Summary:
The behaviour of cells expressing constitutively active HRas is examined in mosaic monolayers, both in MCF10a breast epithelial and Beas2b bronchial epithelial cell lines, mimicking the potential initial phase of development of carcinoma. Single HRas-positive cells are excluded from MCF10a but not Beas2b monolayers. Most interestingly, however, when in groups, these cells are not excluded, but rather sharply segregated within a MCF10a monolayer. In contrast, they freely mix with wt Beas2b cells. Biophysical analysis identifies high tension at heterotypic interfaces between HRas and wild-type cells as the likely reason for segregation of MCF10a cells. The hypothesis is supported experimentally, as myosin inhibition abolishes segregation. The probable reason for the lack of segregation in the bronchial epithelium is to be found in the different intrinsic properties of these cells, which form a looser tissue with lower basal actomyosin activity. The behaviour of single cells and groups is recapitulated in a vortex model based on the principle of differential interfacial tension, under the condition of high heterotypic interfacial tension.
Strengths:
Despite being long recognized as a crucial event during cancer development, segregation of oncogenic cells has been a largely understudied question. This nice work addresses the mechanics of this phenomenon through a straightforward experimental design, applying the biophysical analytical approaches established in the field of morphogenesis. Comparison between two cell types provides some preliminary clues on the diversity of effects in various cancers.
Weaknesses:
Although not calling into question the main message of this study, there are a few issues that one may want to address:
(1) One may be careful in interpreting the comparison between MCF10a and Beas2b cells as used in this study. The conditions may not necessarily be representative of the actual properties of breast and bronchial epithelia. How much of the epithelial organization is reconstituted under these experimental conditions remains to be established. This is particularly obvious for bronchial cells, which would need quite specific culture conditions to build a proper bronchial layer. In this study, they seemed to be on the verge of a mesenchymal phenotype (large gaps, huge protrusions, cells growing on top of each other, as mentioned in the manuscript).
As an alternative to Beas2b, comparison of MCF10a with another cell line capable of more robust in vitro epithelial organization, but ideally with different adhesive and/or tensile properties, would be highly interesting, as it may narrow down the parameters involved in segregation of oncogenic cells.
(2) While the seminal description of tissue properties based on interfacial tensions (Brodland 2002) is clearly key to interpreting these data, the actual "Differential Interfacial Tension Hypothesis" poses that segregation results from global differences, i.e., juxtaposition of two tissues displaying different intrinsic tensions. On the contrary, the results of the present work support a different scenario, where what counts is the actual difference in tension ALONG the tissue boundary, in other words, that segregation is driven by high HETEROTYPIC interfacial tension. This is an important distinction that should be clarified.
(3) Related: The fact that actomyosin accumulates at the heterotypic interface is key here. It would be quite informative to better document the pattern of this accumulation, which is not clear enough from the images of the current manuscript: Are we talking about the actual interface between mutant and wt cells (membrane/cortex of heterotypic contacts)? Or is it more globally overactivated in the whole cell layer along the border? Some better images and some quantification would help.
(4) In the case of Beas2b cells, mutant cells show higher actin than wt cells, while actin is, on the contrary, lower in mutant MCF10a cells (Figure 2b). Has this been taken into account in the model? It may be in line with the idea that HRas may have a different action on the two cell types, a possibility that would certainly be worth considering and discussing.
In conclusion, the study conveys an important message, but, as it stands, the strength of evidence is incomplete. It would greatly benefit from a more detailed and complete analysis of the experimental data, a better fit between this analysis and the corresponding vertex model, and a more in-depth discussion of biological and biophysical aspects. These revisions should be rather easily done, and would then make the evidence much more solid.
-
Reviewer #2 (Public review):
Summary:
The authors investigate the behavior of oncogenic cells in mammary and bronchial epithelia. They observe that individual oncogenic cells are preferentially excluded from the mammary epithelium, but they remain integrated in the bronchial epithelium. They also observe that clusters of oncogenic cells form a compact cluster in the mammary epithelium, but they disperse in the bronchial epithelium. The authors demonstrate experimentally and in the vertex model simulations that the difference in observed behavior is due to the differential tension between the mutant and wild-type cells due to a differential expression of actin and myosin.
Strengths:
(1) Very detailed analysis of experiments to systematically characterize and quantify differences between mammary and bronchial epithelia.
(2) Detailed …
Reviewer #2 (Public review):
Summary:
The authors investigate the behavior of oncogenic cells in mammary and bronchial epithelia. They observe that individual oncogenic cells are preferentially excluded from the mammary epithelium, but they remain integrated in the bronchial epithelium. They also observe that clusters of oncogenic cells form a compact cluster in the mammary epithelium, but they disperse in the bronchial epithelium. The authors demonstrate experimentally and in the vertex model simulations that the difference in observed behavior is due to the differential tension between the mutant and wild-type cells due to a differential expression of actin and myosin.
Strengths:
(1) Very detailed analysis of experiments to systematically characterize and quantify differences between mammary and bronchial epithelia.
(2) Detailed comparison between the experiments and vertex model simulations to identify the differential cell line tension between the oncogenic and wild-type cells as one of the key parameters that are responsible for the different behavior of oncogenic cells in mammary and bronchial epithelia
Weaknesses:
(1) It is unclear what the mechanistic origin of the shape-tension coupling is, which is used in the vertex model, and how important that coupling is for the presented results. The authors claim that the shape-tension coupling is due to the anisotropic distribution of stress fibers when cells are under external stress. It is unclear why the stress fibers should affect an effective line tension on the cell boundaries and why the stress fibers should be sensitive to the magnitude of the internal isotropic cell pressure. In experiments, it makes sense that stress fibers form when cells are stretched. Similar stress fibers form when the cytoskeleton or polymer networks are stretched. It is unclear why the stress fibers should be sensitive to the magnitude of internal isotropic cell pressure. If all the surrounding cells have the same internal pressure, then the cell would not be significantly deformed due to that pressure, and stress fibers would not form. The authors should better justify the use of the shape-tension coupling in the model and also present simulation results without that coupling. I expect that most of the observed behavior is already captured by the differential tension, even if there is no shape-tension coupling.
(2) The observed difference of shape indices between the interfacial and bulk cells in simulations in the absence of differential line tension is concerning. This suggests that either there are not enough statistics from the simulations or that something is wrong with the simulations. For all presented simulation results, the authors should repeat multiple simulations and then present both averages and standard deviations. This way, it would be easier to determine whether the observed differences in simulations are statistically significant.
(3) The authors should also analyze the cell line tension data in simulations and make a comparison with experiments.
-
Author response:
Reviewer #1 (Public review):
Summary:
The behavior of cells expressing constitutively active HRas is examined in mosaic monolayers, both in MCF10a breast epithelial and Beas2b bronchial epithelial cell lines, mimicking the potential initial phase of development of carcinoma. Single HRas-positive cells are excluded from MCF10a but not Beas2b monolayers. Most interestingly, however, when in groups, these cells are not excluded, but rather sharply segregated within a MCF10a monolayer. In contrast, they freely mix with wt Beas2b cells. Biophysical analysis identifies high tension at heterotypic interfaces between HRas and wild-type cells as the likely reason for segregation of MCF10a cells. The hypothesis is supported experimentally, as myosin inhibition abolishes segregation. The probable reason for the lack of …
Author response:
Reviewer #1 (Public review):
Summary:
The behavior of cells expressing constitutively active HRas is examined in mosaic monolayers, both in MCF10a breast epithelial and Beas2b bronchial epithelial cell lines, mimicking the potential initial phase of development of carcinoma. Single HRas-positive cells are excluded from MCF10a but not Beas2b monolayers. Most interestingly, however, when in groups, these cells are not excluded, but rather sharply segregated within a MCF10a monolayer. In contrast, they freely mix with wt Beas2b cells. Biophysical analysis identifies high tension at heterotypic interfaces between HRas and wild-type cells as the likely reason for segregation of MCF10a cells. The hypothesis is supported experimentally, as myosin inhibition abolishes segregation. The probable reason for the lack of segregation in the bronchial epithelium is to be found in the different intrinsic properties of these cells, which form a looser tissue with lower basal actomyosin activity. The behaviour of single cells and groups is recapitulated in a vortex model based on the principle of differential interfacial tension, under the condition of high heterotypic interfacial tension.
Strengths:
Despite being long recognized as a crucial event during cancer development, segregation of oncogenic cells has been a largely understudied question. This nice work addresses the mechanics of this phenomenon through a straightforward experimental design, applying the biophysical analytical approaches established in the field of morphogenesis. Comparison between two cell types provides some preliminary clues on the diversity of effects in various cancers.
Weaknesses:
Although not calling into question the main message of this study, there are a few issues that one may want to address:
(1) One may be careful in interpreting the comparison between MCF10a and Beas2b cells as used in this study. The conditions may not necessarily be representative of the actual properties of breast and bronchial epithelia. How much of the epithelial organization is reconstituted under these experimental conditions remains to be established. This is particularly obvious for bronchial cells, which would need quite specific culture conditions to build a proper bronchial layer. In this study, they seemed to be on the verge of a mesenchymal phenotype (large gaps, huge protrusions, cells growing on top of each other, as mentioned in the manuscript).
We thank the reviewer for this important point. We agree that our experimental conditions do not fully recapitulate the in vivo architecture of either breast or bronchial epithelia. However, here, our intention is to compare two well-established epithelial lines with distinct intrinsic mechanical and organizational properties, rather than to reproduce in-vivo microenvironment. Nevertheless, to address this, we have now strengthened our quantitative analysis of epithelial integrity in Beas2b monolayers, by including ZO-1 immunofluorescence along with E-cadherin immunofluorescence. These measurements confirm that Beas2b monolayers under our culture conditions retain junctional organization, albeit with larger gaps and protrusions compared to MCF10a. We will revise the text to make this distinction explicit.
As an alternative to Beas2b, comparison of MCF10a with another cell line capable of more robust in vitro epithelial organization, but ideally with different adhesive and/or tensile properties, would be highly interesting, as it may narrow down the parameters involved in segregation of oncogenic cells.
We agree with the reviewer that the inclusion of an additional epithelial model system with distinct adhesive and organizational properties would provide valuable insights. In line with this suggestion, we are currently repeating the key experiments using Madin-Darby Canine Kidney (MDCK) cells, a well-established model epithelial cell line. We believe this complementary system will allow us to further dissect the behaviour of HRasV12-expressing cells.
(2) While the seminal description of tissue properties based on interfacial tensions (Brodland 2002) is clearly key to interpreting these data, the actual "Differential Interfacial Tension Hypothesis" poses that segregation results from global differences, i.e., juxtaposition of two tissues displaying different intrinsic tensions. On the contrary, the results of the present work support a different scenario, where what counts is the actual difference in tension ALONG the tissue boundary, in other words, that segregation is driven by high HETEROTYPIC interfacial tension. This is an important distinction that should be clarified.
We thank the reviewer for this insightful comment. As correctly noted, Brodland’s 2002 work provided a seminal formulation of the Differential Interfacial Tension Hypothesis (DITH), which frames tissue organization in terms of effective interfacial tensions. In its original form, DITH emphasized segregation as a consequence of global differences in the intrinsic (bulk) tensions of juxtaposed tissues.
While our results specifically show that segregation is determined by local interfacial mechanics between transformed- and host cells, from our experiments with blebbistatin, where we observed lost in segregation upon reducing global contractility, we believe that the differences in local interfacial mechanics also stem from global differences which belong intrinsically to the tissues in discussion here.
To directly map global interfacial tension, in the revised manuscript, we aim to perform staining with E-cadherin, and actin in the two tissues, and measure cortical actin, stress fibers, and E-cadherin levels at the cell-cell junctions. Once the global tissue mechanics are mapped, we can be more confident about our claim on DITH. Nevertheless, we will also clarify this distinction, more clearly in the text and explicitly state that while DITH provided the foundation for conceptualizing tissue mechanics, our findings on transformed cell- healthy cell interactions specifically demonstrate that segregation is driven by high heterotypic interfacial tension at the tissue boundary.
(3) Related: The fact that actomyosin accumulates at the heterotypic interface is key here. It would be quite informative to better document the pattern of this accumulation, which is not clear enough from the images of the current manuscript: Are we talking about the actual interface between mutant and wt cells (membrane/cortex of heterotypic contacts)? Or is it more globally overactivated in the whole cell layer along the border? Some better images and some quantification would help.
We agree that more detailed visualization of actomyosin distribution would strengthen our conclusion. We are currently working on re-imaging the heterotypic interfaces at higher magnification and are quantifying fluorescence intensity of actin and myosin-II along cell–cell boundaries. All of this will be integrated in the next version of the manuscript.
(4) In the case of Beas2b cells, mutant cells show higher actin than wt cells, while actin is, on the contrary, lower in mutant MCF10a cells (Author response image 2). Has this been taken into account in the model? It may be in line with the idea that HRas may have a different action on the two cell types, a possibility that would certainly be worth considering and discussing.
Our current vertex model does not explicitly incorporate actin levels; rather, it captures their functional consequences indirectly through effective mechanical parameters such as cortical tension and adhesion strength. Nonetheless, we agree that the opposite trends in actin enrichment between Beas2b and MCF10a HRasV12 mutants raise the important possibility that HRas signaling may act through distinct mechanisms in the two cell types.
To further investigate this, we are currently culturing MCF10a and Beas2b HRasV12 mutant populations separately (i.e., without wild-type cells) to assess their intrinsic organization and behavior in isolation. These experiments will help us disentangle how HRas activation differentially impacts epithelial architecture in these two cellular contexts, and we will discuss these ongoing efforts in the revised manuscript.
From the modelling perspective, the model currently does not account for the different actin levels of mutants with respect to wt cells in the two tissues. This can be accounted for by having different and for mutants and wt in the two cases in simulation.
In conclusion, the study conveys an important message, but, as it stands, the strength of evidence is incomplete. It would greatly benefit from a more detailed and complete analysis of the experimental data, a better fit between this analysis and the corresponding vertex model, and a more in-depth discussion of biological and biophysical aspects. These revisions should be rather easily done, and would then make the evidence much more solid.
Reviewer #2 (Public review):
Summary:
The authors investigate the behavior of oncogenic cells in mammary and bronchial epithelia. They observe that individual oncogenic cells are preferentially excluded from the mammary epithelium, but they remain integrated in the bronchial epithelium. They also observe that clusters of oncogenic cells form a compact cluster in the mammary epithelium, but they disperse in the bronchial epithelium. The authors demonstrate experimentally and in the vertex model simulations that the difference in observed behavior is due to the differential tension between the mutant and wild-type cells due to a differential expression of actin and myosin.
Strengths:
(1) Very detailed analysis of experiments to systematically characterize and quantify differences between mammary and bronchial epithelia.
(2) Detailed comparison between the experiments and vertex model simulations to identify the differential cell line tension between the oncogenic and wild-type cells as one of the key parameters that are responsible for the different behavior of oncogenic cells in mammary and bronchial epithelia
Weaknesses:
(1) It is unclear what the mechanistic origin of the shape-tension coupling is, which is used in the vertex model, and how important that coupling is for the presented results. The authors claim that the shape-tension coupling is due to the anisotropic distribution of stress fibers when cells are under external stress. It is unclear why the stress fibers should affect an effective line tension on the cell boundaries and why the stress fibers should be sensitive to the magnitude of the internal isotropic cell pressure. In experiments, it makes sense that stress fibers form when cells are stretched. Similar stress fibers form when the cytoskeleton or polymer networks are stretched. It is unclear why the stress fibers should be sensitive to the magnitude of internal isotropic cell pressure. If all the surrounding cells have the same internal pressure, then the cell would not be significantly deformed due to that pressure, and stress fibers would not form. The authors should better justify the use of the shape-tension coupling in the model and also present simulation results without that coupling. I expect that most of the observed behavior is already captured by the differential tension, even if there is no shape-tension coupling.
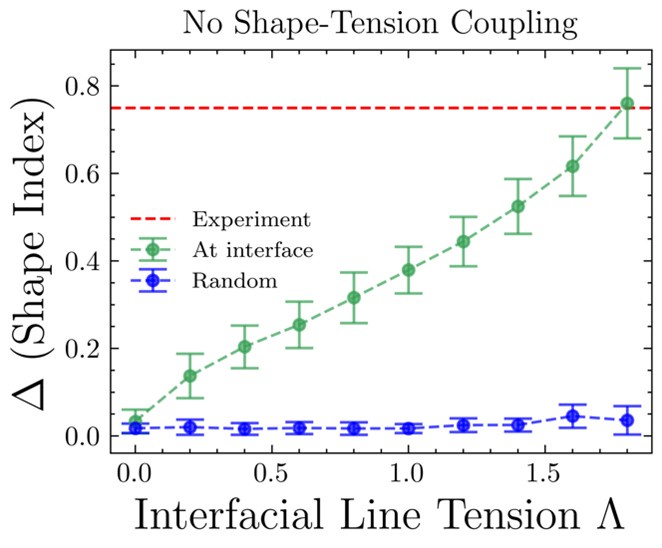
While the segregation behavior can be captured by the differential tension, without the shape-tension coupling, we noticed unjamming and aligned movement of wild type cells at the mutant-cell interface. This was only captured when we incorporated shape tension coupling in the model, suggesting changes in cell shapes due to differential interfacial tension is essential in driving the fate of the mutants. Below, difference between shape indices of cells at the interface and away from the boundary is plotted versus the interfacial tension in the case of no shape-tension coupling [Author response image 1]. The red dashed line represents the experimental value of the shape index difference. The blue line is the shape index difference between two randomly chosen groups of cells (half of the total number of cells in each group is taken). At zero line-tension, the difference in shape index between interface cells and cells away from the interface is same as that between randomly chosen groups of cells, which is expected since there should be no interface at zero line-tension. The no shape-tension data presented here are averaged over 19 seeds. Although the results without shape-tension coupling reaches experimental values at high enough differential tension [Author response image 2], a closer inspection of the simulation results show that the cells are just squeezed and are aligned perpendicular to the interface, which is contrary to what is seen in experiments.
Author response image 1.
Shape indices versus the interfacial line tension
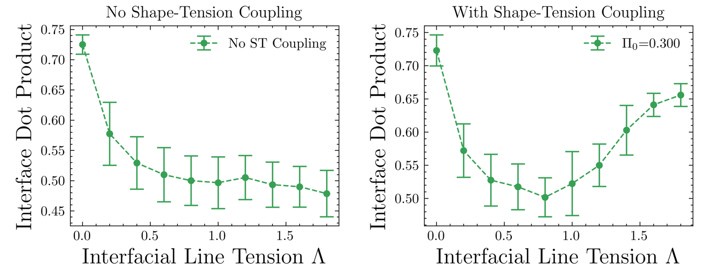
Calculating the average of the absolute value of the dot product of the nematic director and the interface edge for simulations with and without shape-tension coupling clearly shows that with shape-tension coupling, the cells align and elongate along the interface as is seen in experiment, given by an interface dot product value > 0.5 at high enough line-tension values. Further, shape-tension coupling or biased edge tension has been used before to model for cell elongation during embryo elongation [1] and here we use it as an active line-tension force, which elongates cells along the interface, in addition to the differential tension which is passive. This additional quantification of the alignment and elongation of cells along the interface will be added to the Supplementary Information (SI).
[1] Dye, N. A., Popović, M., Iyer, K. V., Fuhrmann, J. F., Piscitello-Gómez, R., Eaton, S., & Jülicher, F. (2021). Self-organized patterning of cell morphology via mechanosensitive feedback. Elife, 10, e57964.
Author response image 2.
Change in interfacial tension with and without shape tension coupling
(2) The observed difference of shape indices between the interfacial and bulk cells in simulations in the absence of differential line tension is concerning. This suggests that either there are not enough statistics from the simulations or that something is wrong with the simulations. For all presented simulation results, the authors should repeat multiple simulations and then present both averages and standard deviations. This way, it would be easier to determine whether the observed differences in simulations are statistically significant.
The reviewer is right in pointing out that statistics for the plots must be shown. The difference in shape indices between the interfacial and bulk cells in simulations has been calculated over 11 different seed values. The observed differences in simulations along with the standard deviations have been plotted below [Author response image 3]. This figure in the paper will be updated to include the standard deviations. The non-zero difference in shape index in the absence of differential line tension for low values of stress threshold is due to the shape-tension coupling acting even at low differential tension. Thus, a non-zero, sufficiently high value of the stress threshold is required in our model with shape-tension coupling, for the model to make sense. This has also been stated in section 4 of the paper. The importance of the stress-tension coupling has been stated in response to the previous point.
Author response image 3.
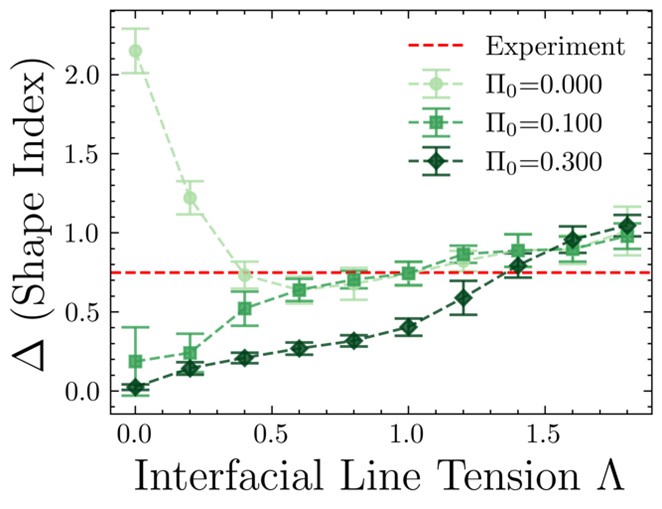
(3) The authors should also analyze the cell line tension data in simulations and make a comparison with experiments.
We agree with the reviewer that cell line tension data should also be analyzed and compared with experiments. This will be added to the next version of the paper.
-

-
-
