Elevated ubiquitin phosphorylation by PINK1 contributes to proteasomal impairment and promotes neurodegeneration
Curation statements for this article:-
Curated by eLife

eLife Assessment
This study provides important insights into the role of polyUbiquitination in neurodegenerative diseases, elucidating how pUb promotes neurodegeneration by affecting proteasomal function. The findings not only offer a new perspective on the pathophysiology of neurodegenerative diseases but also provide potential targets for developing new therapeutic strategies. The results provide solid evidence to support the conclusions.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Ubiquitin (Ub), a central regulator of protein turnover, can be phosphorylated by PINK1 (PTEN-induced putative kinase 1) to generate S65-phosphorylated ubiquitin (pUb). Elevated pUb levels have been observed in aged human brains and in Parkinson’s disease, but the mechanistic link between pUb elevation and neurodegeneration remains unclear. Here, we demonstrate that pUb elevation is a common feature under neurodegenerative conditions, including Alzheimer’s disease, aging, and ischemic injury. We show that impaired proteasomal activity leads to the accumulation of sPINK1, the cytosolic form of PINK1 that is normally proteasome-degraded rapidly. This accumulation increases ubiquitin phosphorylation, which then inhibits ubiquitin-dependent proteasomal activity by interfering with both ubiquitin chain elongation and proteasome-substrate interactions. Specific expression of sPINK1 in mouse hippocampal neurons induced progressive pUb accumulation, accompanied by protein aggregation, proteostasis disruption, neuronal injury, neuroinflammation, and cognitive decline. Conversely, Pink1 knockout mitigated protein aggregation in both mouse brains and HEK293 cells. Furthermore, the detrimental effects of sPINK1 could be counteracted by co-expressing Ub/S65A phospho-null mutant but exacerbated by over-expressing Ub/S65E phospho-mimic mutant. Together, these findings reveal that pUb elevation, triggered by reduced proteasomal activity, inhibits proteasomal activity and forms a feedforward loop that drives progressive neurodegeneration.
Article activity feed
-

-
-
-

eLife Assessment
This study provides important insights into the role of polyUbiquitination in neurodegenerative diseases, elucidating how pUb promotes neurodegeneration by affecting proteasomal function. The findings not only offer a new perspective on the pathophysiology of neurodegenerative diseases but also provide potential targets for developing new therapeutic strategies. The results provide solid evidence to support the conclusions.
-
Reviewer #1 (Public review):
Summary:
The manuscript discusses the role of phosphorylated ubiquitin (pUb) by PINK1 kinase in neurodegenerative diseases. It reveals that elevated levels of pUb are observed in aged human brains and those affected by Parkinson's disease (PD), as well as in Alzheimer's disease (AD), aging, and ischemic injury. The study shows that increased pUb impairs proteasomal degradation, leading to protein aggregation and neurodegeneration. The authors also demonstrate that PINK1 knockout can mitigate protein aggregation in aging and ischemic mouse brains, as well as in cells treated with a proteasome inhibitor. While this study provided some interesting data, several important points should be addressed before being further consideration.
Strengths:
(1) Reveals a novel pathological mechanism of neurodegeneration …
Reviewer #1 (Public review):
Summary:
The manuscript discusses the role of phosphorylated ubiquitin (pUb) by PINK1 kinase in neurodegenerative diseases. It reveals that elevated levels of pUb are observed in aged human brains and those affected by Parkinson's disease (PD), as well as in Alzheimer's disease (AD), aging, and ischemic injury. The study shows that increased pUb impairs proteasomal degradation, leading to protein aggregation and neurodegeneration. The authors also demonstrate that PINK1 knockout can mitigate protein aggregation in aging and ischemic mouse brains, as well as in cells treated with a proteasome inhibitor. While this study provided some interesting data, several important points should be addressed before being further consideration.
Strengths:
(1) Reveals a novel pathological mechanism of neurodegeneration mediated by pUb, providing a new perspective on understanding neurodegenerative diseases.
(2) The study covers not only a single disease model but also various neurodegenerative diseases such as Alzheimer's disease, aging, and ischemic injury, enhancing the breadth and applicability of the research findings.
Comments on revisions:
This study, through a systematic experimental design, reveals the crucial role of pUb in forming a positive feedback loop by inhibiting proteasome activity in neurodegenerative diseases. The data are comprehensive and highly innovative. However, some of the results are not entirely convincing, particularly the staining results in Figure 1.
In Figure 1A, the density of DAPI staining differs significantly between the control patient and the AD patient, making it difficult to conclusively demonstrate a clear increase in PINK1 in AD patients. Quantitative analysis is needed. In Fig 1C, the PINK1 staining in the mouse brain appears to resemble non-specific staining.
-
Author response:
The following is the authors’ response to the previous reviews
In response to Reviewer #1, we have replaced the original images in Figure 1A with new immunofluorescence data showing matched DAPI staining density between control and AD patient samples. We also have updated the PINK1 staining images of mouse brain sections in Figure 1C to eliminate potential non-specific signals. These revisions provide clearer evidence supporting our conclusions about PINK1/pUb’s role in neurodegeneration.
-
-
eLife Assessment
This study provides important insights into the role of polyUbiquitination in neurodegenerative diseases, elucidating how pUb promotes neurodegeneration by affecting proteasomal function. The findings not only offer a new perspective on the pathophysiology of neurodegenerative diseases but also provide potential targets for developing new therapeutic strategies. The experiments in the revised submission provide solid evidence to support the conclusions.
-
Reviewer #1 (Public review):
Summary:
The manuscript discusses the role of phosphorylated ubiquitin (pUb) by PINK1 kinase in neurodegenerative diseases. It reveals that elevated levels of pUb are observed in aged human brains and those affected by Parkinson's disease (PD), as well as in Alzheimer's disease (AD), aging, and ischemic injury. The study shows that increased pUb impairs proteasomal degradation, leading to protein aggregation and neurodegeneration. The authors also demonstrate that PINK1 knockout can mitigate protein aggregation in aging and ischemic mouse brains, as well as in cells treated with a proteasome inhibitor. While this study provided some interesting data, several important points should be addressed before being further consideration.
Strengths:
(1) Reveals a novel pathological mechanism of neurodegeneration …
Reviewer #1 (Public review):
Summary:
The manuscript discusses the role of phosphorylated ubiquitin (pUb) by PINK1 kinase in neurodegenerative diseases. It reveals that elevated levels of pUb are observed in aged human brains and those affected by Parkinson's disease (PD), as well as in Alzheimer's disease (AD), aging, and ischemic injury. The study shows that increased pUb impairs proteasomal degradation, leading to protein aggregation and neurodegeneration. The authors also demonstrate that PINK1 knockout can mitigate protein aggregation in aging and ischemic mouse brains, as well as in cells treated with a proteasome inhibitor. While this study provided some interesting data, several important points should be addressed before being further consideration.
Strengths:
(1) Reveals a novel pathological mechanism of neurodegeneration mediated by pUb, providing a new perspective on understanding neurodegenerative diseases.
(2) The study covers not only a single disease model but also various neurodegenerative diseases such as Alzheimer's disease, aging, and ischemic injury, enhancing the breadth and applicability of the research findings.
Comments on revisions:
This study, through a systematic experimental design, reveals the crucial role of pUb in forming a positive feedback loop by inhibiting proteasome activity in neurodegenerative diseases. The data are comprehensive and highly innovative. However, some of the results are not entirely convincing, particularly the staining results in Figure 1.
In Figure 1A, the density of DAPI staining differs significantly between the control patient and the AD patient, making it difficult to conclusively demonstrate a clear increase in PINK1 in AD patients. Quantitative analysis is needed. In Fig 1C, the PINK1 staining in the mouse brain appears to resemble non-specific staining.
-
Author response:
The following is the authors’ response to the original reviews
Reviewer #1 (Public review):
Summary:
The manuscript discusses the role of phosphorylated ubiquitin (pUb) by PINK1 kinase in neurodegenerative diseases. It reveals that elevated levels of pUb are observed in aged human brains and those affected by Parkinson's disease (PD), as well as in Alzheimer's disease (AD), aging, and ischemic injury. The study shows that increased pUb impairs proteasomal degradation, leading to protein aggregation and neurodegeneration. The authors also demonstrate that PINK1 knockout can mitigate protein aggregation in aging and ischemic mouse brains, as well as in cells treated with a proteasome inhibitor. While this study provided some interesting data, several important points should be addressed before being further considered.
Author response:
The following is the authors’ response to the original reviews
Reviewer #1 (Public review):
Summary:
The manuscript discusses the role of phosphorylated ubiquitin (pUb) by PINK1 kinase in neurodegenerative diseases. It reveals that elevated levels of pUb are observed in aged human brains and those affected by Parkinson's disease (PD), as well as in Alzheimer's disease (AD), aging, and ischemic injury. The study shows that increased pUb impairs proteasomal degradation, leading to protein aggregation and neurodegeneration. The authors also demonstrate that PINK1 knockout can mitigate protein aggregation in aging and ischemic mouse brains, as well as in cells treated with a proteasome inhibitor. While this study provided some interesting data, several important points should be addressed before being further considered.
Strengths:
(1) Reveals a novel pathological mechanism of neurodegeneration mediated by pUb, providing a new perspective on understanding neurodegenerative diseases.
(2) The study covers not only a single disease model but also various neurodegenerative diseases such as Alzheimer's disease, aging, and ischemic injury, enhancing the breadth and applicability of the research findings.
Weaknesses:
(1) PINK1 has been reported as a kinase capable of phosphorylating Ubiquitin, hence the expected outcome of increased p-Ub levels upon PINK1 overexpression. Figures 5E-F do not demonstrate a significant increase in Ub levels upon overexpression of PINK1 alone, whereas the evident increase in Ub expression upon overexpression of S65A is apparent. Therefore, the notion that increased Ub phosphorylation leads to protein aggregation in mouse hippocampal neurons is not yet convincingly supported.
Indeed, overexpression of sPINK1 alone resulted in minimal changes in Ub levels in the soluble fraction (Figure 5E), which is expected given that the soluble Ub pool remains relatively stable and buffered. However, sPINK1* overexpression led to a marked increase in Ub levels in the insoluble fraction, indicative of increased protein aggregation (Figure 5F). The molecular weight distribution of Ub in the insoluble fraction was predominantly below 70 kDa, suggesting that phosphorylation inhibits Ub chain elongation.
To further validate this mechanism, we utilized the Ub/S65A mutant to antagonize Ub phosphorylation and observed a significant reduction in the intensity of aggregated bands at low molecular weights, indicating restored proteasomal activity. The observed increase in Ub levels in the soluble fraction upon Ub/S65A overexpression is likely due to enhanced ubiquitination driven by elevated Ub-S65A, and notably, Ub/S65A was also detectable using an antibody against wild-type Ub.
Consistent with these findings, overexpression of Ub/S65E resulted in a further increase in Ub levels in the insoluble fraction, with intensified low molecular weight bands. The effect was even more pronounced than that observed with sPINK1 transfection, likely resulting from the complete phosphorylation mimicry achieved by Ub/S65E, compared to the relatively low levels of phosphorylation by PINK1.
These findings collectively support the conclusion that sPINK1 promotes protein aggregation via Ub phosphorylation. We have updated the Results and Discussion sections to more clearly present the data and explain the various controls.
(2) The specificity of PINK1 and p-Ub antibodies requires further validation, as a series of literature indicate that the expression of the PINK1 protein is relatively low and difficult to detect under physiological conditions.
We acknowledge the challenges in achieving high specificity with commercially available and customgenerated antibodies targeting PINK1 and pUb, particularly given their low endogenous expression under physiological conditions. However, in our study, we observed robust immunofluorescent staining for PINK1 (Figures 1A, 1C, and 1G) and pUb (Figures 1B, 1D, and 1G) in human brain samples from Alzheimer's disease (AD) patients, as well as in mouse models of AD and cerebral ischemia. The clear visualization can be partly attributed to the pathological upregulation of PINK1 and pUb under disease conditions. Importantly, the images from pink1-/- mice exhibit much weaker staining.
Additionally, we detected a significant elevation in the pUb levels in aged mouse brains compared to younger ones (Figures 1E and 1F). In contrast, pink1-/- mice showed no change in pUb levels with aging, despite some background signals, demonstrating that pUb accumulation during aging is PINK1dependent. Collectively, these results support the specificity of the antibodies used in detecting pathophysiological changes in PINK1 and pUb levels.
For cultured cells, pink1-/- cells served as a negative control for both PINK1 (Figures 2B and 2C) and pUb (Figures 2D and 2E). While the pUb Western blot exhibited some nonspecific background, pUb levels in pink1-/- cells remained unchanged across all MG132 treatment conditions (Figures 2D and 2E), further attesting the usability of the antibodies in conjunction with appropriated controls.
We have updated the manuscript with higher-resolution images; individual image files have been uploaded separately.
(3) In Figure 6, relying solely on Western blot staining and Golgi staining under high magnification is insufficient to prove the impact of PINK1 overexpression on neuronal integrity and cognitive function. The authors should supplement their findings with immunostaining results for MAP2 or NeuN to demonstrate whether neuronal cells are affected.
We included NeuN immunofluorescent staining at 10, 30, and 70 days post transfection in Figure 5— figure supplement 2. The results clearly demonstrate a significant loss of NeuN-positive cells in the hippocampus following Ub/S65E overexpression, while no apparent reduction was observed with sPINK1 transfection alone.
We have also quantified MAP2 protein levels via Western blotting and examined morphology of neuronal dendrite and synaptic structure using Golgi staining. These analyses revealed a significant reduction in MAP2 levels and synaptic damage upon sPINK1 or Ub/S65E overexpression (Figures 6F and 6H), consistent with the proteomics analysis (Figure 5—figure supplementary 5). Notably, these detrimental effects could be rescued by co-expression of Ub/S65A, reinforcing the role of pUb in mediating these structural changes.
Together, our findings from NeuN immunostaining, MAP2 protein analysis, proteomics analysis, and Golgi staining provide strong evidence for the impact of PINK1 overexpression and pUb elevation on neuronal integrity and synaptic structure.
(4) The authors should provide more detailed figure captions to facilitate the understanding of the results depicted in the figures.
Figure captions have been updated with more details incorporated in the revised manuscript.
(5) While the study proposes that pUb promotes neurodegeneration by affecting proteasomal function, the specific molecular mechanisms and signaling pathways remain to be elucidated.
The molecular mechanisms and signaling pathways through which pUb promotes neurodegeneration are likely multifaceted and interconnected. Our findings suggest that mitochondrial dysfunction plays a central role following sPINK1* overexpression. This is supported by (1) an observed increase in full-length PINK1, indicative of impaired mitochondrial quality control, and (2) proteomic data showing enhanced mitophagy at 30 days post-transfection, followed by substantial mitochondrial injuries at 70 days post-transfection (Figure 5—figure supplement 5 and Supplementary Data). The progressive mitochondrial damage caused by protein aggregates would exacerbate neuronal injury and degeneration.
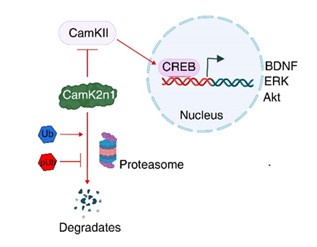
Additionally, reduced proteasomal activity may lead to the accumulation of inhibitory proteins that are normally degraded by the ubiquitin-proteasome system. Our proteomics analysis identified a >50fold increase in CamK2n1 (UniProt ID: Q6QWF9), an endogenous inhibitor of CaMKII activation, following sPINK1* overexpression. The accumulation of CamK2n1 suppresses CaMKII activation, thereby inhibiting the CREB signaling pathway (Figure 7), which is essential for synaptic plasticity and neuronal survival. This disruption can further contribute to neurodegenerative processes.
Thus, our findings underscore the complexity of pUb-mediated neurodegeneration and call for further investigation into downstream consequences.
Reviewer #1 (Recommendations for the authors):
Suggestions for improved or additional experiments, data or analyses.
We have performed additional experiments to investigate how the impairment of ubiquitinproteasomal activity contributes to neurodegeneration. Specifically, we investigated CamK2n1, an endogenous inhibitor of CaMKII, which is normally degraded by the proteasome to allow CaMKII activation. Our proteomics analysis revealed a significant (>50-fold) elevation of CamKI2n1 following sPINK1 overexpression (Figure 5—figure supplement 5 and Supplementary Data).
To validate this mechanism, we conducted immunofluorescence and Western blot analyses, demonstrating reduced levels of phosphorylated CaMKII (pCaMKII) and phosphorylated CREB (pCREB), as well as reduced levels of downstream proteins such as BDNF and ERK. These results have been incorporated into the revised manuscript (Figure 7).
As the proteasome is crucial in maintaining proteostasis, its dysregulation would trigger neurodegeneration through multiple pathways, contributing to a broad cascade of pathological events.
Reviewer #2 (Public review):
Summary:
The manuscript makes the claim that pUb is elevated in a number of degenerative conditions including Alzheimer's Disease and cerebral ischemia. Some of this is based on antibody staining which is poorly controlled and difficult to accept at this point. They confirm previous results that a cytosolic form of PINK1 accumulates following proteasome inhibition and that this can be active. Accumulation of pUb is proposed to interfere with proteostasis through inhibition of the proteasome. Much of the data relies on over-expression and there is little support for this reflecting physiological mechanisms.
Weaknesses:
The manuscript is poorly written. I appreciate this may be difficult in a non-native tongue, but felt that many of the problems are organizational. Less data of higher quality, better controls and incision would be preferable. Overall the referencing of past work is lamentable. Methods are also very poor and difficult to follow.
Until technical issues are addressed I think this would represent an unreliable contribution to the field.
(1) Antibody specificity and detection under pathological conditions
We recognize the limitations of commercially available antibodies for detecting PINK1 and pUb. Nevertheless, our findings reveal a significant elevation in PINK1 and pUb levels under pathological conditions, such as Alzheimer's disease (AD) and ischemia. Additionally, we observed an increase in pUb level during brain aging, further demonstrating its relevance and a potentially causative role for this special pathological condition. Similarly, elevated pUb levels were observed for cultured cells following pharmacological treatment or oxygen-glucose deprivation (OGD).
In contrast, in pink1-/- mice and HEK293 cells used as negative controls, PINK1 and pUb levels remained consistently low. Therefore, the observed elevation of PINK1 and pUb are associated with special pathological conditions, rather than an antibody-detection anomaly.
(2) Overexpression as a model for pathological conditions
To investigate whether the inhibitory effects of sPINK1 on the ubiquitin-proteasome system (UPS) depend on its kinase activity, we employed a kinase-dead version of sPINK1* as a negative control. Given that PINK1 targets multiple substrates, we also investigated whether its effects on UPS inhibition were specifically mediated by ubiquitin phosphorylation. To this end, we used Ub/S65A (a phospho-null mutant) to block Ub phosphorylation by sPINK1, and Ub/S65E (a phospho-mimetic mutant) to mimic phosphorylated Ub. These well-defined controls ensured the robustness of our conclusions.
Although overexpression does not perfectly replicate physiological conditions, it provides a valuable model for studying pathological scenarios such as neurodegeneration and brain aging, where pUb levels are elevated. For example, we observed a 30.4% increase in pUb levels in aged mouse brains compared to young brains (Figure 1F). Similarly, in our sPINK1 overexpression model, pUb levels increased by 43.8% and 59.9% at 30- and 70-days post-transfection, respectively, compared to controls (Figures 5A and 5C). Notably, co-expression of sPINK1* with Ub/S65A almost entirely prevented sPINK1* accumulation (Figure 5B), indicating that an active UPS can efficiently degrade this otherwise stable variant of sPINK1.
Together, our findings demonstrate that sPINK1 accumulation inhibits UPS activity, an effect that can be reversed by the phospho-null Ub mutant. The overexpression model mimics pathological conditions and provides valuable insights into pUb-mediated proteasomal dysfunction.
(3) Organization of the manuscript
Following your suggestion, we have restructured the manuscript to present the key findings in a more logical and cohesive sequence:
(a) Evidence for elevated PINK1 and pUb levels across a broad spectrum of pathological and neurodegenerative conditions;
(b) The effects of pUb elevation in cultured cells, focusing on the proteasome;
(c) Mechanistic insights into how pUb elevation inhibits proteasomal activity;
(d) The absence of PINK1 and pUb alleviates protein aggregation;
(e) Evidence for the causative relationship between elevated pUb levels and proteasomal inhibition;
(f) Demonstration that pUb elevation directly contributes to neuronal degeneration;
(g) Give an additional evidence to explain the mechanism of neuronal degeneration post sPINK1* over-expression. The downstream effects of elevated CamK2n1, an inhibitor of CaMKII, resulting from proteasomal inhibition.
This reorganization should ensure a clear and progressive narrative, and enhance the overall coherence and impact of the revised manuscript.
(4) Revisions to writing, referencing, and methodology
We have made a great effort to enhance the clarity and flow of the manuscript, including the addition of references to appropriately acknowledge prior work. We have also expanded the Methods section with additional details to improve readability and ensure reproducibility. We believe these revisions effectively address the concerns raised and strengthen the overall quality of the manuscript.
Reviewer #2 (Recommendations for the authors):
Figure 1: PINK1 is a poorly expressed protein and difficult to detect by Western blot let alone by immunofluorescence. I have direct experience of the antibody used in this study and do not consider it reliable. There are much cleaner reagents out there, although they still have many challenges. The minimal requirement here is for the PINK1 antibody staining to be compared in wild-type and knockout mice. One would also expect to see a mitochondrial staining which would require higher magnification to be definitive, but it does not look like it to me. This is a key foundational figure and is unreliable. The pUb antibody also has a high background, see for example figure 2E.
Under physiological conditions, PINK1 and pUb levels are indeed low, making their detection challenging. However, under pathological conditions, their expression is significantly elevated, correlating with disease severity. Given the limitations of available reagents, using appropriate controls is a standard approach in biological research.
Nevertheless, we observed robust immunofluorescent staining for PINK1 (Figures 1A, 1C, and 1G) and pUb (Figures 1B, 1D, and 1G) in human brain samples from Alzheimer’s disease (AD) patients and mouse models of AD and cerebral ischemia. Compared to healthy controls, the significant elevation of PINK1 and pUb under these pathological conditions accounts for their clear visualization. To validate antibody specificity, we have included images from pink1-/- mice as negative controls (Figure 1C and 1D, third panel).
Furthermore, we analyzed pUb levels in both young and aged mice, using pink1-/- mice as controls.
Our results revealed a significant increase in pUb levels in aged wild-type mice (Figures 1E and 1F), In contrast, pink1-/- mice exhibited relatively low pUb levels, with no notable change between young and aged groups. These findings reinforce the conclusion that pUb accumulation during aging is dependent on PINK1.Furthermore, we analyzed pUb levels in both young and aged mice, using pink1-/- mice as controls.
For HEK293 cells, pink1-/- cells were used as a negative control for assessing PINK1 (Figures 2B and 2C) and pUb levels (Figures 2D and 2E). While the pUb Western blot did show some nonspecific background, as you have noted, pUb levels significantly increased following MG132 treatment of the wildtype cells. In contrast, no such increase was observed in pink1-/- cells (Figure 2D and 2E). These results further validate the reliability of our findings.
Regarding mitochondrial staining, we recognize that PINK1 localization can vary depending on the pathological context. For example, in Alzheimer’s disease, PINK1 exhibits relatively high nuclear staining, while in cerebral ischemia and brain aging, it is predominantly cytoplasmic and punctate. In contrast, in young, healthy mouse brains, PINK1 is more uniformly distributed. The observed elevation in pUb levels could arise from mitochondrial PINK1 or soluble sPINK1 in the cytoplasm, and it remains unclear whether nuclear PINK1 contributes to pUb accumulation. Investigating the role of PINK1 in different forms and subcellular localizations will be an important avenue for future research.
To enhance clarity, we have updated our images and replaced them with higher-resolution versions in the revised manuscript.
Please also confirm that the GAPDH loading controls represent the same gels, to my eye they do not match.
We have reviewed all the bands, and confirmed that the GAPDH loading controls correspond to the same gels. For different gels, we use separate GAPDH loading controls. There are two experimental scenarios to consider:
(1) When there is a large difference in molecular weight between target proteins, we cut the gel into sections and incubate each section with different antibodies separately.
(2) When the molecular weight difference is small and cutting is not feasible, we first probe the membrane with one antibody, strip it, and then re-incubate the membrane with a second antibody.
These approaches ensure accurate and reliable detection of target proteins with various molecular weights relative to GAPDH.
1H. Ponceau.
We have corrected the spelling.
Figure 2 many elements are confirmation of work already reported and this must be made clearer in the text.
Indeed, the elevation of sPINK1 and pUb upon proteasomal inhibition has been previously reported, and these studies have been acknowledged (Gao, et al, 2016; Dantuma, et al, 2000). In the present study, we expand on these findings by conducting a detailed analysis of the time- and concentrationdependent effects of MG132 on sPINK1 and pUb levels, establishing a causative relationship between pUb accumulation and proteasomal inhibition. Furthermore, we demonstrate that sPINK1 overexpression and MG132-induced proteasomal inhibition exhibit no additive effect, indicating that both converge on the same pathway, resulting in the impairment of proteasomal activity.
It has been established that ubiquitin phosphorylation inhibits Ub chain elongation (Wauer, et al, 2015). However, our study provides novel insights by identifying an additional mechanism: phosphorylated Ub also interferes with the noncovalent interactions between Ub chain and Ub receptors in the proteasome, which further contributes to the impairment of UPS function.
The PINK1 kinase-dead mutant construction (Figure 2F) and the use of Ub-GFP as a proteasomal substrate were based on established methodologies, which have been appropriately cited in the manuscript (Beilina, etal 2005 for KD sPINK1; Yamano, et al for endogenous PINK1; Samant, et al, 2018 and Dantuma, et al, 2000 for Ub-GFP probe). Similarly, our use of puromycin and BALA treatments follows previously reported protocols (Gao, et al, 2016), which allowed us to dissect the relative contributions of sPINK1* overexpression to proteasomal vs. autophagic dysfunction.
As you have noted, our study has built upon prior findings while introducing new mechanistic insights into sPINK1 and pUb-mediated proteasomal dysfunction.
2C 24h MG132 not recommended, most cells are dead by then.
We used MG132 treatment for 24 hours to evaluate the time-course effects of proteasomal inhibition on PINK1 and pUb levels in HEK293 cells (Figures 2C and 2E). We did observe some decrease in both PINK1 and pUb levels at 24 hours compared to 12 hours, which may result from some extend of cell death at the longer treatment duration.
In SH-SY5Y cells, we collected cells at 24 hours after MG132 administration (Figure 5—figure supplementary 1). Though protein aggregation was evident in these cells, we did not observe pronounced cell death under these conditions, justifying our treatment.
Our findings are consistent with previous studies demonstrating that MG132 at 5 µM for 24 hours effectively induces proteasomal inhibition without substantial cytotoxicity. For example, studies using human esophageal squamous cancer cells have reported that this treatment condition inhibits cell proliferation while maintaining cell viability, with cell viability >70% after 24-hour treatment with 5 µM MG132 (Int J Mol Med 33: 1083-1088, 2014).
MG132 has been commonly used at concentrations ranging from 5 to 50 µM for durations of 1 to 24 hours, as stated at the vendor’s website (https://www.cellsignal.com/products/activatorsinhibitors/mg-132/2194).
2I what is BALA do they mean bafilomycin. This is a v-ATPase inhibitor, not just an autophagy inhibitor.
We appreciate the reviewer’s comment regarding the use of BALA in Figure 2I. To clarify, BALA refers to bafilomycin A1, a well-established v-ATPase inhibitor that blocks lysosomal acidification. While bafilomycin A1 is commonly used as an autophagy inhibitor, its primary mechanism involves inhibiting lysosomal function, which is critical for autophagosome-lysosome fusion and subsequent degradation of autophagic cargo.
In our study, we used bafilomycin A1 in conjunction with puromycin to dissect the relative contributions of sPINK1 overexpression on proteasomal and autophagic activities. Puromycin induces protein misfolding and aggregation, causing stress on both degradation pathways. By inhibiting lysosomal function with bafilomycin A1 and blocking the protein degradation load at various stages, we can tell the relative contributions of autophagy and UPS pathways.
We acknowledge that bafilomycin A1’s effects extend beyond autophagy, as it also inhibits v-ATPase activity. However, its inhibition of lysosomal degradation is integral to distinguishing autophagy’s contribution under the experimental conditions, and BALA treatment has been used in extensively in previous studies (Mauvezin and Neufeld, 2015).
We have further clarified this treatment in the revised manuscript.
Figure 3. Legend or text needs to be more explicit about how chains have been produced. From what I can gather from methods only a single E2 has been trialed. Authors should use at least one of the criteria used by Wauer et al. (2014) to confirm the stoichiometry of phosphorylation. The concept that pUb can interfere with E2 discharging is not new, but not universal across E2s.
We have cited in the manuscript that PINK1-mediated ubiquitin phosphorylation can interfere with ubiquitin chain elongation for certain E2 enzymes (Wauer et al., 2015).
To clarify, the focus of our current work is on how elevation of Ub phosphorylation impacts UPS activity, rather than exploring the broader effects of Ub phosphorylation on Ub chain elongation. For this reason, we have used the standard E2 that is well-established for generating K48-linked polyUb chain (Pickart CM, 2005). Moreover, our findings go further and by demonstrate that phosphorylated K48-linked polyubiquitin exhibits weaker non-covalent interactions with proteasomal ubiquitin receptors. This dual effect—on both covalent chain elongation and non-covalent interactions— contributes to the observed reduction in ubiquitin-proteasome activity, a novel aspect of our study.
To address the reviewer’s concerns, we have added details in the Methods section and figure legends regarding the generation of ubiquitin chains. Specifically, we used ubiquitin-activating enzyme E1 (UniProt ID: P22314) and ubiquitin-conjugating enzyme E2-25K (UniProt ID: P61086) to generate K48-linked ubiquitin chains.
Our ESI-MS analysis showed that only 1–2 phosphoryl groups were incorporated into the K48-linked tetra-ubiquitin chains (Figure 3—figure supplement 2). This is consistent with our in vivo findings, where pUb levels increased by 30.4% in aged mouse brains compared to young brains (Figure 1F). Notably, even sub-stoichiometric phosphorylation onto the K48-linked ubiquitin chain significantly weakens the non-covalent interactions with the proteasome (Figures 3E and 3H).
Figure 4. I could find no definition of the insoluble fraction, nor details on how it is prepared.
The insoluble fraction primarily contains proteins that are aggregated or associated with hydrophobic interactions and cannot be solubilized by RIPA buffer. We have provided more details in the Methods of the revised manuscript about how the insoluble fraction was prepared. Our approach was based on established protocols for fractionating soluble and insoluble proteins from brain tissues (Wirths, 2017). Here is an outline of the procedure, which enables the separation and subsequent analysis of distinct protein populations:
• Lysis and preparation of soluble fraction: Cells and brain tissues were lysed using RIPA buffer (Beyotime Biotechnology, cat# P0013B) containing protease (P1005) and phosphatase inhibitors (P1081) on ice for 30 minutes, with gentle vortexing every 10 minutes. Brain samples were homogenized using a precooled TissuePrep instrument (TP-24, Gering Instrument Company). Lysates were centrifuged at 12,000 rpm for 30 minutes at 4°C. The supernatant was collected as the soluble protein fraction.
• Preparation of insoluble fraction: The pellet was resuspended in 20 µl of SDS buffer (2% SDS, 50 mM Tris-HCl, pH 7.5) and subjected to ultrasonic pyrolysis at 4°C for 8 cycles (10 seconds ultrasound, 30 seconds interval). The samples were then centrifuged at 12,000 rpm for 30 minutes at 4°C. The supernatant obtained after this step was designated as the insoluble protein fraction.
• Protein quantification: Protein concentrations for both soluble and insoluble fractions were determined using the BCA Protein Assay Kit (Beyotime Biotechnology, cat# P0009).
Figure 5. What is the transfection efficiency? How many folds is sPINK1 over-expressed? Typically, a neuron will have only a few hundred copies of PINK1 at the basal state. How much mutant ubiquitin is expressed relative to wild type, seeing the free ubiquitin signals on the gels might be helpful here, but they seem to have been cut off.
We appreciate the reviewer's insightful comments regarding transfection efficiency, the extent of sPINK1 overexpression, and the expression levels of mutant ubiquitin relative to wild-type ubiquitin. Below, we provide detailed responses to each point:
Transfection Efficiency: Our immunofluorescent staining for NeuN, a neuronal marker, demonstrated that over 90% of NeuN-positive cells were co-localized with GFP (Figure 5—figure supplement 2), indicating a high transfection efficiency in our neuronal cultures.
Extent of sPINK1 Overexpression: Quantifying the exact fold increase of sPINK1 upon overexpression is inherently difficult due to its low basal expression under physiological conditions, making the relative increase difficult to measure (small denominator effect). However, our Western blot analysis shows that ischemic events can cause a substantial elevation of PINK1 levels, including both full-length and cleaved forms (Figure 1H). This suggests that our overexpression model recapitulates the pathological increase in PINK1, making it a relevant system for studying disease mechanisms.
From Figure 5B, it is evident that sPINK1 levels differ significantly between neurons overexpressing sPINK1 alone and those co-expressing sPINK1 + Ub/S65A (70 days post-transfection). Overexpression of sPINK1 alone results in multiple PINK1 bands, consistent with sPINK1, endogenous PINK1 (induced by mitochondrial damage), and ubiquitinated sPINK1. In comparison, co-expressing Ub/S65A leads to faint PINK1 bands, suggesting that in the presence of a functionally restored proteasome, overexpressed sPINK1 is rapidly degraded. Therefore, actual accumulation of sPINK1 depends on proteasomal activity, and the “over-expressed” PINK1 level can be comparable to levels observed under native, pathological conditions.
Expression Levels of Mutant Ubiquitin Relative to Wild-Type: Assessing the expression levels of mutant versus wild-type ubiquitin is indeed valuable. In Figure 5E, we observed a 38.9% increase in high-molecular-weight ubiquitin conjugates in the soluble fraction when comparing the sPINK1+Ub/S65A group to the control. This increase suggests that mutant ubiquitin is actively incorporated into polyubiquitin chains.
Regarding free monomeric ubiquitin, its low abundance and rapid incorporation into polyubiquitin chains make it difficult to visualize in Western blots. Additionally, its low molecular weight and lower antibody binding valency further reduce its visibility.
General: a number of effects are shown following over-expression but no case is made that these levels of pUb are ever attained physiologically. I am very unconvinced by these findings and think the manuscript needs to be improved at multiple levels before being added to the record.
We understand the reviewer’s concerns regarding the relevance of pUb levels observed in our overexpression model. To clarify, our study is not focused on physiological levels of pUb, but rather on pathologically elevated levels, which have been documented in various neurodegenerative conditions. While overexpression is not a perfect replication of pathological states, it provides a valuable tool to investigate mechanisms that become relevant under disease conditions. Moreover, we have taken steps to ensure the validity of our findings and to address potential limitations associated with overexpression models:
Pathological Relevance: Besides several reported literatures, we observed significant increases in PINK1 and pUb levels in human brain samples from Alzheimer's disease (AD) patients, as well as in mouse models of AD, cerebral ischemia (including mouse middle cerebral artery occlusion ischemic model and oxygen glucose deprivation cell model), and aging (e.g., Figures 1E, 1F, and 1H). All these data show that pUb levels are elevated under pathological conditions. Our overexpression model mimics these pathological scenarios by recreating the high levels of pUb, which lead to the impairment of proteasomal activity and subsequent disruption of proteostasis.
Use of Robust Controls: To ensure the reliability of our results and interpretations, we employed multiple controls for our experiments. We have used pink1-/- mice and cells to confirm that pUb accumulation is PINK1-dependent (Figures 1C and 2C). We have also included kinase-dead sPINK1 mutant and Ub/S65A phospho-null mutants to negate/counteract the specific roles of PINK1 activity and pUb in proteasomal dysfunction. On the other hand, we have used Ub/S65E for phosphomimetic mutant, corresponding to a 100% Ub phosphorylation.
Importantly, we have compared sPINK1 overexpression with both baseline and disease-mimicking conditions, thus to ensure that the observed effects are consistent with pathological changes. Furthermore, our findings are supported by complementary evidences from human brain samples, model animals, cell cultures, and molecular assays. Integrating the different controls and various approaches, we have provided mechanistic insights into how elevated pUb levels causes proteasomal impairment and contributes to neurodegeneration.
Our findings elucidate how elevated pUb level contributes to the disruption of proteostasis in neurodegenerative conditions. While overexpression may have limitations, it remains a powerful tool for dissecting pathological mechanisms and testing hypotheses. Our results align with and expand upon previous studies suggesting pUb as a biomarker of neurodegeneration (Hou, et al, 2018; Fiesel, et al, 2015), and provide mechanistic insights into how elevated pUb and sPINK1 drive a viscous feedforward cycle, ultimately leading to proteasomal dysfunction and neurodegeneration.
We hope these clarifications highlight the relevance and rigor of our study, and welcome additional suggestions to improve the manuscript.
Reviewer #3 (Public review):
Summary:
This study aims to explore the role of phosphorylated ubiquitin (pUb) in proteostasis and its impact on neurodegeneration. By employing a combination of molecular, cellular, and in vivo approaches, the authors demonstrate that elevated pUb levels contribute to both protective and neurotoxic effects, depending on the context. The research integrates proteasomal inhibition, mitochondrial dysfunction, and protein aggregation, providing new insights into the pathology of neurodegenerative diseases.
Strengths:
- The integration of proteomics, molecular biology, and animal models provides comprehensive insights.
- The use of phospho-null and phospho-mimetic ubiquitin mutants elegantly demonstrates the dual effects of pUb.
- Data on behavioral changes and cognitive impairments establish a clear link between cellular mechanisms and functional outcomes.
Weaknesses:
- While the study discusses the reciprocal relationship between proteasomal inhibition and pUb elevation, causality remains partially inferred.
It has been well-established that protein aggregates, particularly neurodegenerative fibrils, can impair proteasomal activity (McDade, et al., 2024; Kinger, et al., 2024; Tseng, et al., 2008). Other contributing factors, including ATP depletion, reduced proteasome component expression, and covalent modifications of proteasomal subunits, can also lead to declined proteasomal function. Additionally, mitochondrial injury serves as an important source of elevated PINK1 and pUb levels. Recent studies have demonstrated that efficient mitophagy is essential to prevent pUb accumulation, whereas partial mitophagy failure results in elevated PINK1 levels (Chin, et al, 2023; Pollock, et al. 2024).
While pathological conditions can impair proteasomal function and slow sPINK1 degradation, leading to its accumulation, our results demonstrate that overexpression of sPINK1 or PINK1 can initiate this cycle as well. Once this cycle is initiated, it becomes self-perpetuating, as sPINK1 and pUb accumulation progressively impair proteasomal function, leading to more protein aggregates and mitochondrial damages.
Importantly, we show that co-expression of Ub/S65A effectively rescues cells from this cycle, which further illustrates the pivotal role of pUb in driving proteasomal inhibition and the causality between pUb elevation and proteasomal inhibition. At the animal level, pink1 knockout prevents protein aggregation under aging and cerebral ischemia conditions (Figures 1E and 1G).
Together, by controlling at protein, cell, and animal levels, our findings support this self-reinforcing and self-amplifying cycle of pUb elevation, proteasomal inhibition, protein aggregation, mitochondrial damage, and ultimately, neurodegeneration.
- The role of alternative pathways, such as autophagy, in compensating for proteasomal dysfunction is underexplored.
Indeed, previous studies have shown that elevated sPINK1 can enhance autophagy (Gao, et al., 2016,), potentially compensating for impaired UPS function. One mechanism involves PINK1mediated phosphorylation of p62, which enhances autophagic activity.
In our study, we observed increased autophagic activity upon sPINK1 overexpression, as shown in Figure 2I (middle panel, without BALA). This increase in autophagy may facilitate the degradation of ubiquitinated proteins induced by puromycin, partially mitigating proteasomal dysfunction. This compensation might also explain why protein aggregation, though statistically significant, increased only slightly at 70 days post-sPINK1 transfection (Figure 5F). Additionally, we detected a mild but statistically insignificant increase in LC3II levels in the hippocampus of mouse brains at 70 days postsPINK1 transfection (Figure 5—figure supplement 6), further supporting the notion of autophagy activation.
However, while autophagy may provide some compensation, its effect is likely limited. The UPS and autophagy serve distinct roles in protein degradation:
• Autophagy is a bulk degradation pathway, primarily targeting damaged organelles, intracellular pathogens, and protein aggregates, often in a non-selective manner.
• The UPS, in contrast, is highly selective, degrading short-lived regulatory proteins, misfolded proteins, and proteins tagged for degradation via ubiquitination.
Thus, while sPINK1 overexpression enhances autophagy-mediated degradation, it simultaneously impairs UPS-mediated degradation. This suggests that autophagy partially compensates for proteasomal dysfunction but is insufficient to counterbalance the UPS's selective degradation function. We have incorporated additional discussion in the revised manuscript.
- The immunofluorescence images in Figure 1A-D lack clarity and transparency. It is not clear whether the images represent human brain tissue, mouse brain tissue, or cultured cells. Additionally, the DAPI staining is not well-defined, making it difficult to discern cell nuclei or staging. To address these issues, lower-magnification images that clearly show the brain region should be provided, along with improved DAPI staining for better visualization. Furthermore, the Results section and Figure legends should explicitly indicate which brain region is being presented. These concerns raise questions about the reliability of the reported pUb levels in AD, which is a critical aspect of the study's findings.
We have taken steps to address the concerns regarding clarity and transparency in Figure 1A-D. We have already addressed the source of tissues at the left of each images. For example, we have written “human brain with AD” at the left side of Figure 1A, and “mouse brains with AD” at the left side of Figure 1C.
Briefly, the human brain samples in Figure 1 originate from the cingulate gyrus of Alzheimer’s disease (AD) patients. Our analysis revealed that PINK1 is primarily localized within cell bodies, whereas pUb is more abundant around Aβ plaques, likely in nerve terminals. For the mouse brain samples, we have now explicitly indicated in the figure legends and Results section that the images represent the neocortex of APP/PS1 mice, a mouse model relevant to AD pathology, as well as the corresponding regions in wild-type and pink1-/- mice. We have ensured that the brain regions and sources are clearly stated throughout the manuscript.
Regarding image clarity, we have uploaded higher-resolution versions of the images in the revised manuscript to improve visualization of key features, including DAPI staining. We believe these revisions enhance the reliability and interpretability of our findings, particularly in relation to the reported pUb levels in AD.
- Figure 4B should also indicate which brain region is being presented.
The images were taken for layer III-IV in the neocortex of mouse brains. We have included this information in the figure legend of the revised manuscript.
Reviewer #3 (Recommendations for the authors):
- Expand on the potential compensatory role of autophagy in response to proteasomal dysfunction.
Upon proteasomal inhibition, cells may activate autophagy as an alternative pathway of degradation to help clear damaged or misfolded proteins. Autophagy is a bulk degradation process that targets long-lived proteins, damaged organelles, and aggregated proteins for lysosomal degradation. While this pathway can provide some compensation, it is distinct from the ubiquitin-proteasome system (UPS), which specializes in the selective degradation of short-lived regulatory proteins and misfolded proteins.
In our study, we observed increased autophagic activity following sPINK1 overexpression (Figure 2J, middle panel, without BALA) and a slight, though statistically insignificant, increase in LC3II levels in the hippocampus of mouse brains at 70 days post-sPINK1 transfection (Figure 5—figure supplement 6). These findings suggest that autophagy is indeed upregulated as a compensatory response to proteasomal dysfunction, potentially facilitating the degradation of aggregated ubiquitinated proteins. Additionally, gene set enrichment analysis (GSEA) revealed similar enrichment of autophagy pathways at 30 and 70 days post-sPINK1 overexpression (Figure 5—figure supplement 5).
However, the compensatory capacity of autophagy is likely limited. While autophagy can reduce protein aggregation, it is an inherently non-selective process and cannot fully replace the targeted functions of the UPS. Moreover, as we illustrate in Figure 7 of the revised manuscript, UPS is essential for degrading specific regulatory and inhibitory proteins and plays a critical role in cellular proteostasis, particularly in signaling regulation, cell cycle control, and stress responses.
Together, while autophagy activation provides some degree of compensation, it cannot fully restore cellular proteostasis. The interplay between these two degradation pathways is an important area for future investigation. For the present study, our focus is on how pUb elevations impact proteasomal activity and elicits downstream effects.
We have incorporated these additional discussions on this topic in the revised manuscript.
- Simplify the discussion of complex mechanisms to improve accessibility for readers.
We have revised the Discussion to present the mechanisms in a more coherent and accessible manner, ensuring clarity for a broader readership. These revisions should make the discussion more intuitive while preserving the depth of our findings.
- Statistical analyses could benefit from clarifying how technical replicates and biological replicates were accounted for across experiments.
We have clarified our statistical analysis in the Methods section and figure legends, explicitly detailing how many biological replicates were accounted for across experiments. These revisions should enhance transparency and clarity, ensuring that our findings are robust and reproducible.
- The image in Figure 3D is too small to distinguish any signals. A larger and clearer image should be presented.
We have expanded the images in Figure 3D. Additionally, we have replaced figures with version of better resolutions throughout the manuscript.
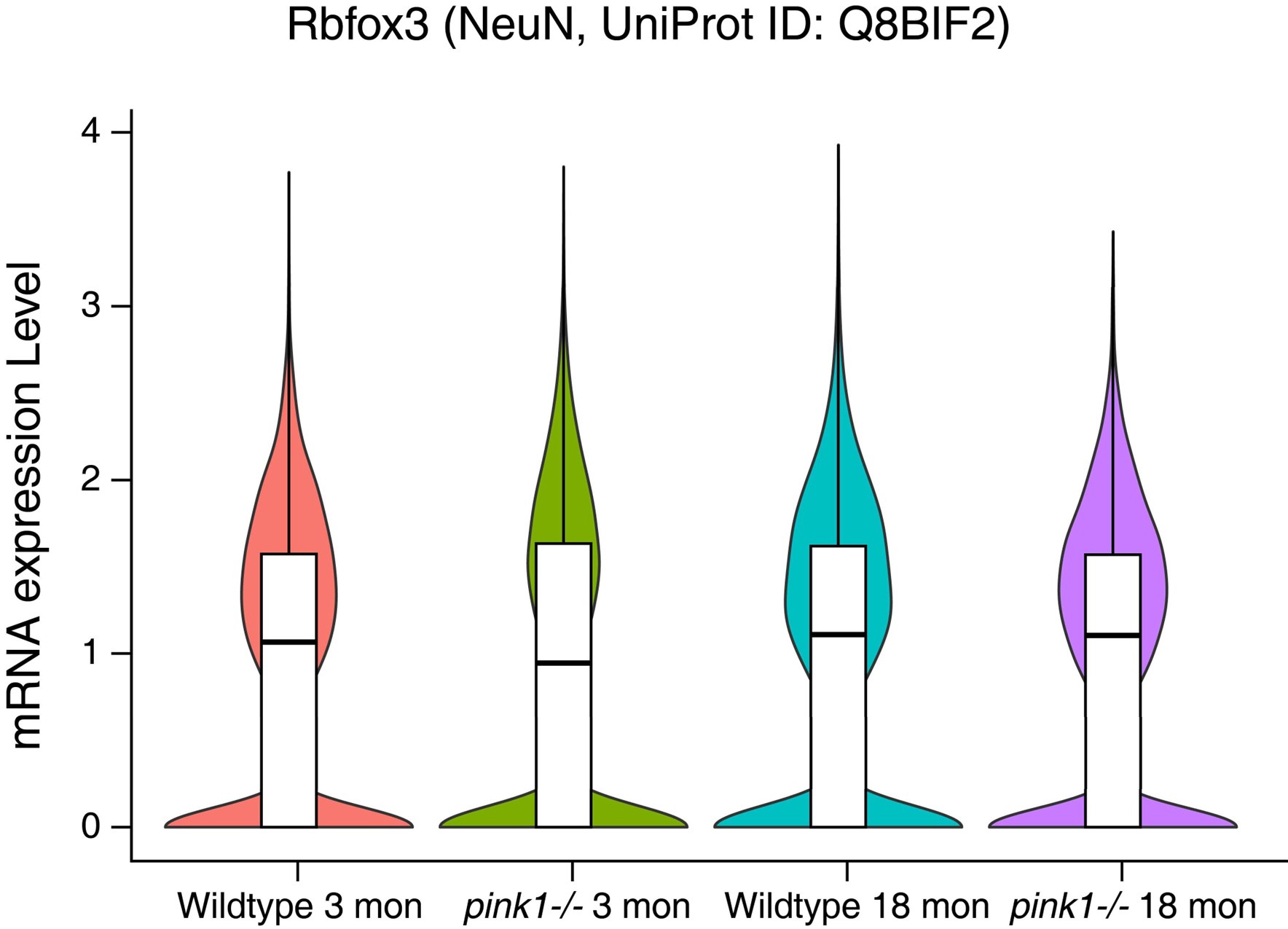
- NeuN expression in Figure 4B differs between wildtype and pink-/- mice. Additional validation is needed to determine whether pink-/- enhances NeuN expression.
The difference in NeuN immunofluorescence intensity between wild-type and pink1-/- mice in Figure 4B may simply result from variations in image acquisition rather than an actual difference in NeuN expression.
Our single nuclei RNA-seq analyses of wild-type and pink1-/- mice at 3 and 18 months of age reveal no significant differences in NeuN expression at the transcript level (data provided below). This confirms that the observed variation in fluorescence intensity is unlikely to reflect an authentic upregulation of NeuN expression. Thus, factors like the concentration of antibody, image exposure and processing may contribute to differences in staining intensity.
Author response image 1.
-
Author response:
Public Reviews:
Reviewer #1 (Public review):Summary:
The manuscript discusses the role of phosphorylated ubiquitin (pUb) by PINK1 kinase in neurodegenerative diseases. It reveals that elevated levels of pUb are observed in aged human brains and those affected by Parkinson's disease (PD), as well as in Alzheimer's disease (AD), aging, and ischemic injury. The study shows that increased pUb impairs proteasomal degradation, leading to protein aggregation and neurodegeneration. The authors also demonstrate that PINK1 knockout can mitigate protein aggregation in aging and ischemic mouse brains, as well as in cells treated with a proteasome inhibitor. While this study provided some interesting data, several important points should be addressed before being further considered.
Strengths:
(1) Reveals a novel pathological …
Author response:
Public Reviews:
Reviewer #1 (Public review):Summary:
The manuscript discusses the role of phosphorylated ubiquitin (pUb) by PINK1 kinase in neurodegenerative diseases. It reveals that elevated levels of pUb are observed in aged human brains and those affected by Parkinson's disease (PD), as well as in Alzheimer's disease (AD), aging, and ischemic injury. The study shows that increased pUb impairs proteasomal degradation, leading to protein aggregation and neurodegeneration. The authors also demonstrate that PINK1 knockout can mitigate protein aggregation in aging and ischemic mouse brains, as well as in cells treated with a proteasome inhibitor. While this study provided some interesting data, several important points should be addressed before being further considered.
Strengths:
(1) Reveals a novel pathological mechanism of neurodegeneration mediated by pUb, providing a new perspective on understanding neurodegenerative diseases.
(2) The study covers not only a single disease model but also various neurodegenerative diseases such as Alzheimer's disease, aging, and ischemic injury, enhancing the breadth and applicability of the research findings.
Weaknesses:
(1) PINK1 has been reported as a kinase capable of phosphorylating Ubiquitin, hence the expected outcome of increased p-Ub levels upon PINK1 overexpression. Figures 5E-F do not demonstrate a significant increase in Ub levels upon overexpression of PINK1 alone, whereas the evident increase in Ub expression upon overexpression of S65A is apparent. Therefore, the notion that increased Ub phosphorylation leads to protein aggregation in mouse hippocampal neurons is not yet convincingly supported.
Indeed, overexpression of sPINK1* alone caused little change in Ub levels in the soluble fraction (Figure 5E), which is expected. Ub in the soluble fraction is in a relatively stable, buffered state. However, overexpression of sPINK1* resulted in an increase in Ub levels in the insoluble fraction, indicating protein aggregation. The molecular weight of Ub in the insoluble fraction was predominantly below 70 kDa, implying that phosphorylation inhibits Ub chain elongation.
To further examine this, we used the Ub/S65A mutant to antagonize Ub phosphorylation, and found that the aggregation at low molecular weight was significantly reduced, indicating a partial restoration of proteasomal activity. The increase in Ub levels in both the soluble and insoluble fractions likely results from the high rate of ubiquitination driven by the elevated levels of Ub. Notably, the overexpressed Ub/S65A was detected in the Western blot using the wild-type Ub antibody, which accounts for the apparently increased Ub level.
When overexpressing Ub/S65E, we again saw an increase in Ub levels in the insoluble fraction (but no increase in the soluble fraction), with low molecular weight bands even more prominent than those observed with sPINK1* transfection. These findings collectively support the conclusion that sPINK1* promotes protein aggregation through Ub phosphorylation.
(2) The specificity of PINK1 and p-Ub antibodies requires further validation, as a series of literature indicate that the expression of the PINK1 protein is relatively low and difficult to detect under physiological conditions.
We acknowledge the challenges in achieving optimal specificity for commercially available and custom-generated antibodies targeting PINK1 and pUb, particularly given the low endogenous levels of these proteins under physiological conditions. Despite these limitations, we observed robust immunofluorescent staining for PINK1 (Figures 1A, 1C, and 1G) and pUb (Figures 1B, 1D, and 1G) in human brain samples from Alzheimer's disease (AD) patients, as well as in mouse brains from models of AD and cerebral ischemia. The significant elevation of PINK1 and pUb under these pathological conditions likely accounts for the clear visualization. To validate antibody specificity, we have included images from pink1-/- mice as negative controls in the revised manuscript (Figure 1C and 1D, third panel).
In addition, we detected a significant increase in pUb levels in aged mouse brains compared to young ones (Figures 1E and 1F). Notably, in pink1-/- mice, pUb levels remained unchanged between young and aged groups, despite some background signal, further supporting the conclusion that pUb accumulation during aging is PINK1-dependent.
In HEK293 cells, pink1-/- cells served as a negative control for PINK1 (Figure 2B and 2C) and for pUb (Figure 2D and 2E). While the Western blot using the pUb antibody displayed some nonspecific background, pUb levels in pink1-/- cells remained unchanged across all MG132 treatment conditions (Figures 2D and 2E), further attesting the reliability of our findings.
(3) In Figure 6, relying solely on Western blot staining and Golgi staining under high magnification is insufficient to prove the impact of PINK1 overexpression on neuronal integrity and cognitive function. The authors should supplement their findings with immunostaining results for MAP2 or NeuN to demonstrate whether neuronal cells are affected.
Thank you for raising this important point. We included NeuN immunofluorescent staining in Figure 5—figure supplement 2 of the original manuscript. The results demonstrate a significant loss of NeuN-positive cells in the hippocampus following Ub/S65E overexpression, while no apparent change in NeuN-positive cells was observed with sPINK1* transfection alone. These findings provide evidence of neuronal loss in response to Ub/S65E, further supporting the impact of pUb elevation on neuronal integrity.
While we did not perform MAP2 immunostaining, we included complementary analyses to assess neuronal integrity. Specifically, we performed Western blotting to determine MAP2 protein levels and used Golgi staining to study neuronal morphology and synaptic structure in greater detail. These analyses revealed that overexpression of sPINK1* or Ub/S65E decreased MAP2 levels and caused damage to synaptic structures (Figures 6F and 6H). Importantly, the deleterious effects of sPINK1* overexpression could be rescued by co-expression of Ub/S65A, further underscoring the role of pUb in mediating these changes.
Together, our NeuN immunostaining, MAP2 analysis, and Golgi staining provide strong evidence for the impact of PINK1 overexpression and pUb elevation on neuronal integrity and synaptic health. We believe these complementary approaches sufficiently address the reviewer’s concern and highlight the pathological consequences of elevated pUb levels.
(4) The authors should provide more detailed figure captions to facilitate the understanding of the results depicted in the figures.
Figure captions will be updated with more details in the revised manuscript.
(5) While the study proposes that pUb promotes neurodegeneration by affecting proteasomal function, the specific molecular mechanisms and signaling pathways remain to be elucidated.
The specific molecular mechanisms and signaling pathways through which pUb promotes neurodegeneration are likely multifaceted and interconnected. Mitochondrial dysfunction appears to be a central contributor to neurodegeneration following sPINK1* overexpression. This is supported by (1) an observed increase in full-length PINK1, indicative of impaired mitochondrial quality control, and (2) proteomic data revealing enhanced mitophagy at 30 days post-transfection and substantial mitochondrial injury by 70 days post-transfection. The progressive damage to mitochondria caused by protein aggregates can cause further neuronal injury and degeneration.
In addition, reduced proteasomal activity may result in the accumulation of inhibitory proteins that are normally degraded by the ubiquitin-proteasome system. Our proteomics analysis identified a >54-fold increase in CamK2n1 (UniProt ID: Q6QWF9), an endogenous inhibitor of CaMKII activation, following sPINK1* overexpression. This is particularly significant because the accumulation of CamK2n1 could suppress CaMKII activation and, subsequently, inhibit the CREB signaling pathway (illustrated below). As CREB is essential for synaptic plasticity and neuronal survival, its inhibition may further amplify neurodegenerative processes.
While our study identifies proteasomal dysfunction and mitochondrial damage as key initial triggers, downstream effects—such as disruptions in signaling pathways like CaMKII-CREB—likely contribute to a broader cascade of pathological events. These findings highlight the complexity of pUb-mediated neurodegeneration and suggest that further exploration of downstream mechanisms is necessary to fully elucidate the pathways involved.
We plan to include the proteomics data, in the revised manuscript, of mouse brain tissues at 30 days and 70 days post-transfection, to further highlight this downstream effect upon proteasomal dysfunction.
Author response image 1.
Reviewer #2 (Public review):
Summary:
The manuscript makes the claim that pUb is elevated in a number of degenerative conditions including Alzheimer's Disease and cerebral ischemia. Some of this is based on antibody staining which is poorly controlled and difficult to accept at this point. They confirm previous results that a cytosolic form of PINK1 accumulates following proteasome inhibition and that this can be active. Accumulation of pUb is proposed to interfere with proteostasis through inhibition of the proteasome. Much of the data relies on over-expression and there is little support for this reflecting physiological mechanisms.
Weaknesses:
The manuscript is poorly written. I appreciate this may be difficult in a non-native tongue, but felt that many of the problems are organisational. Less data of higher quality, better controls and incision would be preferable. Overall the referencing of past work is lamentable.
Methods are also very poor and difficult to follow.
Until technical issues are addressed I think this would represent an unreliable contribution to the field.(1) Antibody specificity and detection under pathological conditions
We acknowledge the limitations of commercially available antibodies for detecting PINK1 and pUb. Despite these challenges, our findings demonstrate a significant increase in PINK1 and pUb levels under pathological conditions, such as Alzheimer's disease (AD) and ischemia. Additionally, we observed an increase in pUb level during brain aging, further highlighting its relevance in this particular physiological process. To ensure reliable quantification of PINK1 and pUb levels, we used pink1-/- mice and HEK293 cells as negative controls. For example, PINK1 levels were extremely low in control cells but increased dramatically after 2 hours of oxygen-glucose deprivation (OGD) and 6 hours of reperfusion (Figure 1H). Together, these controls validate that the observed elevations in PINK1 and pUb are specific and linked to pathological or certain physiological conditions.
(2) Overexpression as a model for pathological conditions
To investigate whether the inhibitory effects of sPINK1* on the ubiquitin-proteasome system (UPS) are dependent on its kinase activity, we utilized a kinase-dead version of sPINK1* as a negative control. Since PINK1 has multiple substrates, we further explored whether its effects on UPS inhibition were mediated specifically by ubiquitin phosphorylation. For this, we used Ub/S65A (a phospho-null mutant) to antagonize Ub phosphorylation by sPINK1*, and Ub/S65E (a phospho-mimetic mutant) to mimic phosphorylated Ub. These well-defined controls ensured the robustness of our conclusions.
While overexpression does not perfectly replicate physiological conditions, it serves as a valuable model for studying pathological scenarios such as neurodegeneration and brain aging, where pUb levels are known to increase. For example, we observed a 30.4% increase in pUb levels in aged mouse brains compared to young brains (Figure 1F). Similarly, in our sPINK1* overexpression model, pUb levels increased by 43.8% and 59.9% at 30- and 70-days post-transfection, respectively, compared to controls (Figures 5A and 5C). Notably, co-expression of sPINK1* with Ub/S65A almost entirely prevented sPINK1* accumulation (Figure 5B), indicating that an active UPS can efficiently degrade sPINK1*. Collectively, these findings show that sPINK1* accumulation inhibits UPS activity, a defect that can be rescued by the phospho-null Ub mutant. Thus, this overexpression model closely mimics pathological conditions and offers valuable insights into pUb-mediated proteasomal dysfunction.
(3) Organization of the manuscript
We believe the structure of the manuscript is justified and systematically addresses the key aspects of the study in a logic flow:
(a) Evidence for the increase of PINK1 and pUb in multiple pathological and physiological conditions.
(b) Identification of the sources and consequences of sPINK1 and pUb elevation.
(c) Mechanistic insights into how pUb inhibits UPS-mediated degradation.
(d) Validation of these findings using pink1-/- mice and cells.
(e) Evidence of the reciprocal relationship between proteasomal inhibition and pUb elevation, culminating in neurodegeneration.
(f) Demonstration of elevated pUb levels and protein aggregation in the hippocampus following sPINK1* overexpression, supported by proteomic analyses, behavioral tests, Western blotting, and Golgi staining.
Thus, this organization provides a clear and cohesive narrative, culminating in the demonstration that sPINK1* overexpression induces hippocampal neuron degeneration.
(4) Revisions to writing, referencing, and methodology
We will improve the clarity and flow of the manuscript, add more references to properly acknowledge prior work, and incorporate additional details into the Methods section to enhance readability and reproducibility. These improvements should address the organizational and technical concerns raised, while strengthen the overall quality of the manuscript.
Reviewer #3 (Public review):
Summary:
This study aims to explore the role of phosphorylated ubiquitin (pUb) in proteostasis and its impact on neurodegeneration. By employing a combination of molecular, cellular, and in vivo approaches, the authors demonstrate that elevated pUb levels contribute to both protective and neurotoxic effects, depending on the context. The research integrates proteasomal inhibition, mitochondrial dysfunction, and protein aggregation, providing new insights into the pathology of neurodegenerative diseases.
Strengths:
- The integration of proteomics, molecular biology, and animal models provides comprehensive insights.
- The use of phospho-null and phospho-mimetic ubiquitin mutants elegantly demonstrates the dual effects of pUb.
- Data on behavioral changes and cognitive impairments establish a clear link between cellular mechanisms and functional outcomes.
Weaknesses:
- While the study discusses the reciprocal relationship between proteasomal inhibition and pUb elevation, causality remains partially inferred.
The reciprocal cycle between proteasomal inhibition and pUb elevation can be initiated by various factors that impair proteasomal activity. These factors include Aβ accumulation, ATP depletion, reduced expression of proteasome components, and covalent modifications of proteasomal subunits—all well-established contributors to the progressive decline in proteasome function. Once initiated, this cycle would become self-perpetuating, with the accumulation of sPINK1 and pUb driving a feedback loop of deteriorating proteasomal activity.
In the current study, this reciprocal relationship between sPINK1/pUb elevation and proteasomal dysfunction is depicted in Figure 4A. Our results demonstrate that increased sPINK1 or PINK1 levels, such as through overexpression, can initiate this cycle. Crucially, co-expression of Ub/S65A effectively rescues the cells from this cycle, highlighting the pivotal role of pUb in driving proteasomal inhibition and establishing causality in this relationship. At the animal level, pink1 knockout could prevent protein aggregation upon aging and cerebral ischemia (Figures 1E and 1G).
Mitochondrial injury is a likely source of elevated PINK1 and pUb levels. A recent study showed that efficient mitophagy is necessary to prevent pUb accumulation (bioRxiv 2023.02.14.528378), suggesting that mitochondrial damage can trigger this cycle. In another study (bioRxiv 2024.07.03.601901), the authors found that mitochondrial damage could enhance PINK1 transcription, further increasing cytoplasmic PINK1 levels and exacerbating the cycle.
- The role of alternative pathways, such as autophagy, in compensating for proteasomal dysfunction is underexplored.
Elevated sPINK1 has been reported to enhance autophagy (Autophagy 2016, 12: 632-647), potentially compensating for the impaired UPS. One mechanism involves the phosphorylation of p62 by sPINK1, which enhances autophagy activity. In our study, we did observe increased autophagic activity upon sPINK1* overexpression, as shown in Figure 2I (middle panel, without BALA). This increased autophagy may help degrade ubiquitinated proteins induced by puromycin, partially compensating for the proteasomal dysfunction.
This compensation might explain why protein aggregation only increased slightly, though statistically significant, at 70 days post sPINK1* transfection (Figure 5F). Additionally, we observed a slight, though statistically insignificant, increase in LC3II levels in the hippocampus of mouse brains at 70 days post sPINK1* transfection (Figure 5—figure supplement 6), further supporting the notion of autophagy activation.
However, while autophagy may provide some compensation, its effect is likely limited. Autophagy and UPS differ significantly in their roles and mechanisms of degradation. Autophagy is a bulk degradation pathway that is generally non-selective, targeting long-lived proteins, damaged organelles, and intracellular pathogens. In contrast, the UPS is highly selective, primarily degrading short-lived regulatory proteins, misfolded proteins, and proteins tagged for degradation.
Together, we found that sPINK1* overexpression enhanced autophagy-mediated protein degradation while simultaneously impairing UPS-mediated degradation. This suggests that while autophagy may provide partial compensation for proteasomal dysfunction, it is not sufficient to fully counterbalance the selective degradation functions of the UPS.
- The immunofluorescence images in Figure 1A-D lack clarity and transparency. It is not clear whether the images represent human brain tissue, mouse brain tissue, or cultured cells. Additionally, the DAPI staining is not well-defined, making it difficult to discern cell nuclei or staging. To address these issues, lower-magnification images that clearly show the brain region should be provided, along with improved DAPI staining for better visualization. Furthermore, the Results section and Figure legends should explicitly indicate which brain region is being presented. These concerns raise questions about the reliability of the reported pUb levels in AD, which is a critical aspect of the study's findings.
We will include low-magnification images in the supplementary figures of the revised manuscript to provide a broader context for the immunofluorescence data presented in Figure 1. DAPI staining at higher magnifications will also be provided to improve visualization of cell nuclei and overall tissue structure. Additionally, we will indicate the brain regions examined in the corresponding figure legends, and incorporate more details in the Results section to provide clearer descriptions of the samples and brain regions analyzed.
The human brain samples presented in Figure 1 are from the cingulate gyrus region of Alzheimer's disease (AD) patients. Our analysis revealed that PINK1 is primarily localized within cell bodies, while pUb is more abundant around Aβ plaques, likely in nerve terminals. These additional clarifications and supplementary figures should provide greater transparency and improve the reliability of our findings.
- Figure 4B should also indicate which brain region is being presented.
The images were taken for layer III-IV in the neocortex of mouse brains, which information will be incorporated in the figure legend of the revised manuscript.
-
eLife Assessment
The useful manuscript presents interesting findings in the field of neurodegenerative diseases by highlighting the dual role of phosphorylated ubiquitin (pUb) in cellular proteostasis and neurotoxicity. However, some claims for discovery are supported by unconvincing and incomplete evidence that requires further validation. The poor quality of key immunofluorescent images and questionable quantification analysis raise technical concerns.
-
Reviewer #1 (Public review):
Summary:
The manuscript discusses the role of phosphorylated ubiquitin (pUb) by PINK1 kinase in neurodegenerative diseases. It reveals that elevated levels of pUb are observed in aged human brains and those affected by Parkinson's disease (PD), as well as in Alzheimer's disease (AD), aging, and ischemic injury. The study shows that increased pUb impairs proteasomal degradation, leading to protein aggregation and neurodegeneration. The authors also demonstrate that PINK1 knockout can mitigate protein aggregation in aging and ischemic mouse brains, as well as in cells treated with a proteasome inhibitor. While this study provided some interesting data, several important points should be addressed before being further considered.
Strengths:
(1) Reveals a novel pathological mechanism of neurodegeneration …
Reviewer #1 (Public review):
Summary:
The manuscript discusses the role of phosphorylated ubiquitin (pUb) by PINK1 kinase in neurodegenerative diseases. It reveals that elevated levels of pUb are observed in aged human brains and those affected by Parkinson's disease (PD), as well as in Alzheimer's disease (AD), aging, and ischemic injury. The study shows that increased pUb impairs proteasomal degradation, leading to protein aggregation and neurodegeneration. The authors also demonstrate that PINK1 knockout can mitigate protein aggregation in aging and ischemic mouse brains, as well as in cells treated with a proteasome inhibitor. While this study provided some interesting data, several important points should be addressed before being further considered.
Strengths:
(1) Reveals a novel pathological mechanism of neurodegeneration mediated by pUb, providing a new perspective on understanding neurodegenerative diseases.
(2) The study covers not only a single disease model but also various neurodegenerative diseases such as Alzheimer's disease, aging, and ischemic injury, enhancing the breadth and applicability of the research findings.Weaknesses:
(1) PINK1 has been reported as a kinase capable of phosphorylating Ubiquitin, hence the expected outcome of increased p-Ub levels upon PINK1 overexpression. Figures 5E-F do not demonstrate a significant increase in Ub levels upon overexpression of PINK1 alone, whereas the evident increase in Ub expression upon overexpression of S65A is apparent. Therefore, the notion that increased Ub phosphorylation leads to protein aggregation in mouse hippocampal neurons is not yet convincingly supported.
(2) The specificity of PINK1 and p-Ub antibodies requires further validation, as a series of literature indicate that the expression of the PINK1 protein is relatively low and difficult to detect under physiological conditions.
(3) In Figure 6, relying solely on Western blot staining and golgi staining under high magnification is insufficient to prove the impact of PINK1 overexpression on neuronal integrity and cognitive function. The authors should supplement their findings with immunostaining results for MAP2 or NeuN to demonstrate whether neuronal cells are affected.
(4) The authors should provide more detailed figure captions to facilitate the understanding of the results depicted in the figures.
(5) While the study proposes that pUb promotes neurodegeneration by affecting proteasomal function, the specific molecular mechanisms and signaling pathways remain to be elucidated. -
Reviewer #2 (Public review):
Summary:
The manuscript makes the claim that pUb is elevated in a number of degenerative conditions including Alzheimer's Disease and cerebral ischaemia. Some of this is based on antibody staining which is poorly controlled and difficult to accept at this point. They confirm previous results that a cytosolic form of PINK1 accumulates following proteasome inhibition and that this can be active. Accumulation of pUb is proposed to interfere with proteostasis through inhibition of the proteasome. Much of the data relies on over-expression and there is little support for this reflecting physiological mechanisms.
Weaknesses:
The manuscript is poorly written. I appreciate this may be difficult in a non-native tongue, but felt that many of the problems are organisational. Less data of higher quality, better controls …
Reviewer #2 (Public review):
Summary:
The manuscript makes the claim that pUb is elevated in a number of degenerative conditions including Alzheimer's Disease and cerebral ischaemia. Some of this is based on antibody staining which is poorly controlled and difficult to accept at this point. They confirm previous results that a cytosolic form of PINK1 accumulates following proteasome inhibition and that this can be active. Accumulation of pUb is proposed to interfere with proteostasis through inhibition of the proteasome. Much of the data relies on over-expression and there is little support for this reflecting physiological mechanisms.
Weaknesses:
The manuscript is poorly written. I appreciate this may be difficult in a non-native tongue, but felt that many of the problems are organisational. Less data of higher quality, better controls and incision would be preferable. Overall the referencing of past work is lamentable.
Methods are also very poor and difficult to follow.Until technical issues are addressed I think this would represent an unreliable contribution to the field.
-
Reviewer #3 (Public review):
Summary:
This study aims to explore the role of phosphorylated ubiquitin (pUb) in proteostasis and its impact on neurodegeneration. By employing a combination of molecular, cellular, and in vivo approaches, the authors demonstrate that elevated pUb levels contribute to both protective and neurotoxic effects, depending on the context. The research integrates proteasomal inhibition, mitochondrial dysfunction, and protein aggregation, providing new insights into the pathology of neurodegenerative diseases.
Strengths:
- The integration of proteomics, molecular biology, and animal models provides comprehensive insights.
- The use of phospho-null and phospho-mimetic ubiquitin mutants elegantly demonstrates the dual effects of pUb.
- Data on behavioral changes and cognitive impairments establish a clear link …Reviewer #3 (Public review):
Summary:
This study aims to explore the role of phosphorylated ubiquitin (pUb) in proteostasis and its impact on neurodegeneration. By employing a combination of molecular, cellular, and in vivo approaches, the authors demonstrate that elevated pUb levels contribute to both protective and neurotoxic effects, depending on the context. The research integrates proteasomal inhibition, mitochondrial dysfunction, and protein aggregation, providing new insights into the pathology of neurodegenerative diseases.
Strengths:
- The integration of proteomics, molecular biology, and animal models provides comprehensive insights.
- The use of phospho-null and phospho-mimetic ubiquitin mutants elegantly demonstrates the dual effects of pUb.
- Data on behavioral changes and cognitive impairments establish a clear link between cellular mechanisms and functional outcomes.Weaknesses:
- While the study discusses the reciprocal relationship between proteasomal inhibition and pUb elevation, causality remains partially inferred.
- The role of alternative pathways, such as autophagy, in compensating for proteasomal dysfunction is underexplored.
- The immunofluorescence images in Figure 1A-D lack clarity and transparency. It is not clear whether the images represent human brain tissue, mouse brain tissue, or cultured cells. Additionally, the DAPI staining is not well-defined, making it difficult to discern cell nuclei or staging. To address these issues, lower-magnification images that clearly show the brain region should be provided, along with improved DAPI staining for better visualization. Furthermore, the Results section and Figure legends should explicitly indicate which brain region is being presented. These concerns raise questions about the reliability of the reported pUb levels in AD, which is a critical aspect of the study's findings.
- Figure 4B should also indicate which brain region is being presented. -
-
